The emergence of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that causes respiratory coronavirus disease 2019 (COVID-19) triggered an unprecedented public health emergency. Human mobility patterns were fundamental to the spread of SARS-CoV-2 globally. The situation is particularly acute in a country like Saudi Arabia, which has seen more than 750,000 cases and over 9,000 deaths as of March 30th, 2022.
Mass religious gatherings frequently occur in the kingdom, causing major population movements. However, due to public health measures, major outbreaks have been avoided for the most part in recent years. During the Hajj and Umrah, roughly 9.5 million pilgrims visit two Islamic sites in Makkah and Madinah annually. Additionally, more than 5 million Shiite Saudi nationals travel to Iran for pilgrimage, contributing to the origin of SARS-CoV-2 infections in the region. A clear indication of this movement is the early phase of the COVID-19 epidemic within Saudi Arabia, where the first case was officially reported on March 2nd, 2020, in Qatif.
Frequent mutations of the virus render newer variants, which are constantly emerging. Genomic epidemiology of the variants is a valuable tool for investigating outbreaks and viral progress. It is essential to monitor the genetic diversity of the virus as the newer variants can functionally impact infection transmission and severity.
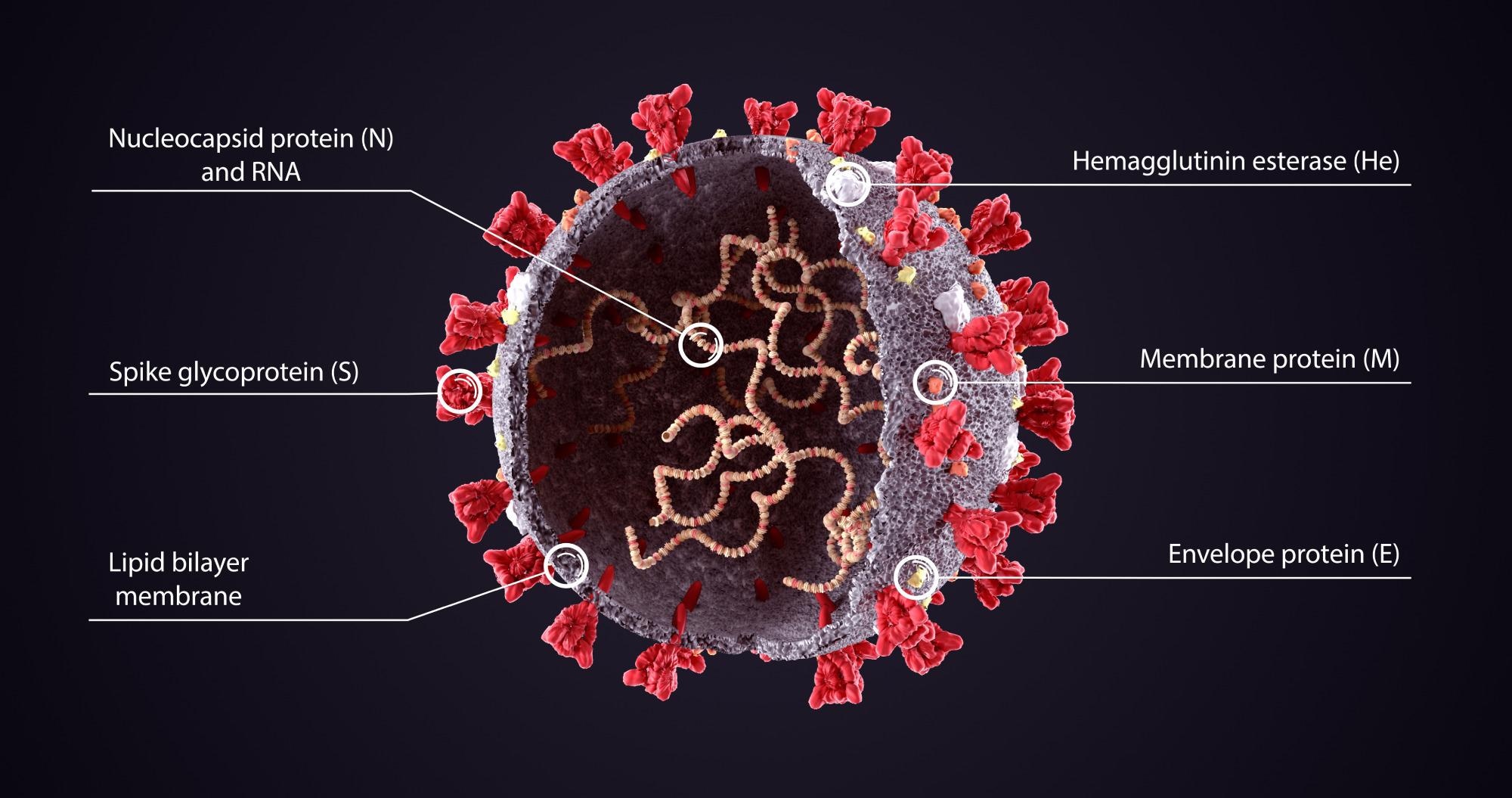 Study: SARS-CoV-2 genomes from Saudi Arabia implicate nucleocapsid mutations in host response and increased viral load. Image Credit: Orpheus FX / Shutterstock
Study: SARS-CoV-2 genomes from Saudi Arabia implicate nucleocapsid mutations in host response and increased viral load. Image Credit: Orpheus FX / Shutterstock
The study
A recent study published in Nature Communications journal analyzed SARS-CoV-2 genomes for nucleotide changes, mutations, genetic diversity, and the nature of transmission. Specific mutations were linked to viral loads, and their impact on virus-host interactions was assessed.
The study entailed sequencing 892 genomes of the virus derived from nasopharyngeal swab samples of patients in Saudi Arabia between March-August 2020.
![a Locations of the sampling cities within Saudi Arabia. b Stacked bars showing the numbers of samples retrieved from the 4 cities and the Eastern region during the first six months of the pandemic. Cities are colored as in panel a. Months are shown at the bottom of the figure, and each month is divided into 5-day intervals. New daily cases for the city of Khobar are shown on the Eastern Region plot. Major restrictions imposed by the Ministry of Health and by Royal decrees are indicated above plots. c Stacked bars showing the average numbers of new daily cases in sampling cities (Supplementary Note 1). d Estimate of effective reproduction number [Rt] over time in Saudi Arabia (top) and the estimate of effective population size [Ne], the relative population size required to produce the diversity seen in the sample (bottom). Central black lines show median estimates, and gray confidence areas denote the 95% credible intervals. The red horizontal red line represents an R of 1, the level required to sustain epidemic growth.](https://www.news-medical.net/images/news/ImageForNews_708976_16486101544482053.png)
a Locations of the sampling cities within Saudi Arabia. b Stacked bars showing the numbers of samples retrieved from the 4 cities and the Eastern region during the first six months of the pandemic. Cities are colored as in panel a. Months are shown at the bottom of the figure, and each month is divided into 5-day intervals. New daily cases for the city of Khobar are shown on the Eastern Region plot. Major restrictions imposed by the Ministry of Health and by Royal decrees are indicated above plots. c Stacked bars showing the average numbers of new daily cases in sampling cities (Supplementary Note 1). d Estimate of effective reproduction number [Rt] over time in Saudi Arabia (top) and the estimate of effective population size [Ne], the relative population size required to produce the diversity seen in the sample (bottom). Central black lines show median estimates, and gray confidence areas denote the 95% credible intervals. The red horizontal red line represents an R of 1, the level required to sustain epidemic growth.
Results
Out of the sequenced 892 SARS-CoV-2 genomes, 836 single-nucleotide polymorphisms (SNP) relative to the Wuhan-Hu-1 isolate were detected, which is generally lower than the global samples. In addition, a total of 41 insertion/deletion polymorphisms (indels) were found, of which 26 were detected in the coding regions. Indels were specific to a sample with a maximum of four samples with identical indels.
Samples from Saudi Arabia revealed a higher frequency of SNPs relative to the global SNP data, including the Spike protein D614G and other three successive SNPs generating the R203K and G204R changes amidst the nucleocapsid protein. It was inferred that the effective reproduction number [R(t)] decreased with time until late June; April 27th, 2020, was the decline's starting point. According to the analysis, viral diversity peaked early in the month of June.
Five major Nextstrain clades (19A-B and 20A-C) were detected from the collected samples. Clade 20A with nucleocapsid protein (N) mutations (R203K/G204R) were associated with higher incidences of hospitalization in the intensive care unit (ICU). Clade 19B was probably imported in late February 2020, whereas the other clades were imported approximately in March 2020.
Clades 20A-C were predominated by R203K/G204R SNPs. These SNPs are also found in early presenting 19A and 19B. The frequency of R203K/G204R peaked in Saudi Arabia earlier compared to the global peak. Globally, the initial peak of R203K/G204R SNPs was observed in July 2020, a gradual decline during fall 2020, and another increase in the SNPs and an increased Spike mutation Y501N was found in the B1.1.17 lineage.
Disease severity was defined by the number of patients admitted to the intensive care unit (ICU) and deaths. In addition, higher viral load was associated with Spike protein D614G SNP resulting from A23403 mutation.
For assessing the severity and mortality, 12 SNPs together with R203K/G204R SNPs were included, and a significant association was found between the severity of the disease and R203K/G204R SNPs. C14408T and C1887T SNP showed a positive association and C241T SNP revealed a negative association.
A positive association was found between R203K/G204R SNPs and mortality in the model that excluded time. Other SNPs did not show any significant association. If time was included as a variable, no association was observed between R203K/G204R SNPs and mortality.
A statistically significant positive relation was found between viral copy number and R203K/G204R, A23403G, and C26735T SNPs, whereas a negative association was observed for C14408T, C3037T, and G25563T SNPs.
N-protein of SARS-CoV-2, central to viral replication, binds the viral RNA-genome. R203K/G204R mutations destabilize the N structure and enhance the protein-RNA binding, altering the protein's response to events concerning serine phosphorylation. R203K/G204R mutations in N protein are associated with serine/arginine-rich motif-containing linkage region linked to oligomerization of N proteins.
At low protein concentrations, control N protein has lower oligomerization potential compared to mutant N protein. A strong binding between viral ribonucleic acid (RNA) and mutant N protein was detected.
Overall, 43 human proteins were identified that significantly and differentially interacted with the control and mutant N protein – one with decreased interaction and 42 showing increased interactions with the mutant N protein. Among the 42 human proteins, proteins associated with viral transcription, signaling pathways, viral processes, apoptosis, and cell death, and negative regulatory RNA nuclear export were identified. Additionally, mutant proteins associated with translation and immune processes were identified.
Ribosomal subunit export and negatively regulated transfer RNA (tRNA) were found to be associated with the most enhanced biological processes suggesting an efficient inhibition of the viral pathogenesis and replication by the mutant virus. Many viruses could enhance pathogenesis and survival by exploiting the host sumoylation process. The antiviral mechanism and sumoylation pathways were also identified.
High phosphorylation was detected at the serine 206 sites in the mutant N protein. Both mutant and control N proteins revealed serine site phosphorylation within the linkage region (LKR).
In addition, 153 and 144 differentially expressed (DE) genes were identified in N-mutant and N-control transfected cells. After fold-change adjustment, interferon-related genes revealed a vigorous overexpression in the N-mutant transfected cells. Overexpression of STAT1, TMPRSS13, and ACE2 genes were also found.
Virus-associated overexpression of biological processes was noted in analyzing the up-regulated genes. Differentially expressed genes were associated with substantially enriched pathways like cytokine production, interferon-related response, and viral reproductive processes. The relation between these pathways and up-regulated overlapping genes was displayed.
It can be inferred that R203K/G204R mutations provoke an overexpression of the interferon-related genes contributing to the cytokine storm aggravating the COVID-19 pathogenesis.
Therefore, R203K/G204R mutations in N-protein can increase the potency of SARS-CoV-2 infection and the host response. The present study identified pathways that could be potential targets for future therapies.
Journal reference:
- Mourier, T., Shuaib, M., Hala, S., et al. (2022). SARS-CoV-2 genomes from Saudi Arabia implicate nucleocapsid mutations in host response and increased viral load. Nature Communications. doi: 10.1038/s41467-022-28287-8. https://www.nature.com/articles/s41467-022-28287-8
 Excess tubulin disrupts tissue organization and cell survival
Excess tubulin disrupts tissue organization and cell survival