The coronavirus disease-19 (COVID-19) pandemic that emerged in Wuhan, China, in late December 2019 spread rapidly throughout the world and has caused over 6.5 million deaths. The disease is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a positive sense RNA virus that can lead to various clinical symptoms causing the hospitalization of many people with pneumonitis. Research has highlighted that COVID-19 patients have a high arterial and venous thrombosis rate involving pulmonary embolism, deep-vein thrombosis, ischemic stroke, and myocardial infarction. In addition, many postmortem examinations indicated the presence of microthrombi in the heart, lungs, brain, liver, and kidney of COVID-19 patients. This suggests that COVID-19 can cause systemic thrombosis, leading to multi-organ failure.
Previous studies have also observed that COVID-19 patients have a high level of serum coagulation markers such as D-dimer and fibrinogen. In addition, Thromboprophylaxis has been observed to improve outcomes in hospitalized COVID-19 patients. However, it did not significantly affect the mortality of patients with severe COVID-19. Therefore, thrombosis remains a prominent COVID-19 feature where high thrombin generation is not the only contributing factor.
Platelets essential for hemostasis can also lead to thrombosis due to inappropriate activation. Platelets can become hyperactive, leading to the increased secretion of dense alpha-granules, increased formation of platelet-leukocyte aggregates, and increased aggregation. Platelet transcriptome analysis has shown an overrepresentation of mitochondrial dysfunction and antigen presentation pathways in COVID-19 patients, leading to platelet hyperactivity. However, many studies also reported reduced or impaired platelet function in COVID-19 patients, suggesting that the platelet response is complex.
Although the pathogenesis of thrombosis in COVID-19 patients is not well understood, it comprises several hallmarks of thromboinflammation. SARS-CoV-2 can activate endothelial cells with the help of the angiotensin-converting enzyme 2 (ACE2) receptor, which leads to vascular dysfunction. Endothelium damage can activate the innate immune system through neutrophil recruitment, tissue factor (TF) expression, pro-inflammatory cytokines, and complement. This leads to higher expression and/or release of prothrombotic factors and upregulation of adhesion molecules which can activate platelets, causing thrombus formation, aggregation, and further activation of the innate immune system. Platelets can then increase the generation of thrombin through the expression of phosphatidyl serine and tissue factor (TF). This network of interactions between the innate immune system, platelets, coagulation, and damaged endothelium can contribute to thrombosis in COVID-19 patients.
A new study posted in the pre-print server bioRxiv* aimed to analyze the impact of COVID-19 on platelet proteome and relate the functional responses of platelets and the formation of platelet-neutrophil aggregate in hospitalized COVID-19 patients.
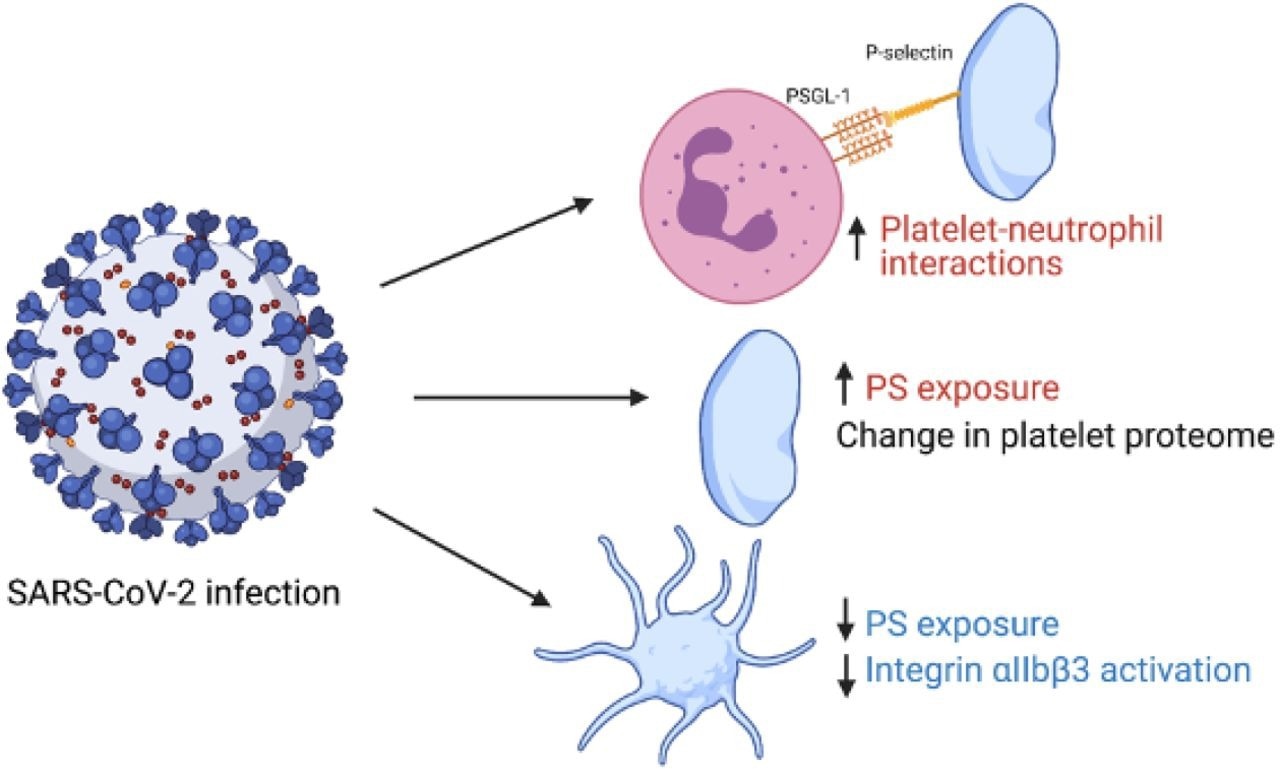 Graphical summary of study findings. Red indicates an increase in variable in patients with COVID- 19 vs healthy controls and blue indicates a decrease. Figure made using biorender.com.
Graphical summary of study findings. Red indicates an increase in variable in patients with COVID- 19 vs healthy controls and blue indicates a decrease. Figure made using biorender.com.
About the study
The study involved 22 hospitalized adult COVID-19 patients and 19 healthy controls recruited between October 2020 and February 2021. Information on comorbidities, demographics, medications, admission results and treatment records were collected from all the patients. Moreover, full blood counts were measured for both healthy controls and COVID-19 patients. Platelets were isolated from venous blood, washed, and prepared for tandem mass tag (TMT) proteomic analysis.
The platelet proteome was compared between healthy controls and COVID-19 patients. In addition, Gene ontology pathways were used to analyze COVID-19 elated changes to platelet proteins that affect their function. Finally, western blotting and flow cytometry analysis was carried out.
Study findings
The results indicated that the mean age of the COVID-19 cohort was 59 years, and the control cohort was 39 years. The mean body mass index (BMI) of COVID-19 patients was reported to be 30.6 kg/m2, while that of the control was 22.7 kg/m2. In addition, 88% of the patients required oxygen therapy and additional medications, including rivaroxaban, dexamethasone, and heparin. Additionally, COVID-19 patients were observed to comprise a higher neutrophil level and lower lymphocyte count.
Out of the 5,773 platelet proteins, 858 were elevated in COVID-19 patients compared to healthy controls. These mainly included the C-reactive protein and the 40S ribosomal proteins. However, many platelet activations signaling proteins decreased in COVID-19 patients compared to controls. Platelet lysates obtained from COVID-19 patients were reported to show a reduction in thrombopoietin (TPO) receptor cMpl, interferon-induced transmembrane membrane protein 3 (IFITM3), and protein kinase C α (PKCα) expression. Additionally, CD147 (basigin), a receptor associated with SAS-CoV-2 interaction, was reported to be present in both control and COVID-19 patient samples.
Out of the 38 serum proteins associated with COVID-19, 12 were altered in the platelet of COVID-19 patients. Out of them, four proteins were observed to show more than a 4-fold expression that included serum amyloid A (SAA1), galectin 3-binding protein (G3BP), lipopolysaccharide-binding protein (LBP), and C-reactive protein (CRP). Out of 18 granule secretion proteins associated with COVID-19, 11 were observed to be increased, and seven decreased in platelets from COVID-19 patients. Moreover, out of the 15 platelet activation proteins, 12 were reduced in COVID-19 patients' platelets, including Ser/Thr kinases and tyrosine kinases. However, levels of interleukin-6 receptor subunit, Apolipoprotein E, and cathepsin G were observed to be increased.
Activation of integrin αIIbβ3, which is the receptor causing platelet aggregation and alpha-granule marker P-selectin that is expressed on the secretion of alpha-granule secretion, was reported to be similar for both controls and COVID-19 patients under basal conditions. However, agonist-induced integrin αIIbβ3 activation was reported to be impaired in platelets of COVID-19 patients, while agonist-stimulated P-selectin expression was reported to be unchanged. Additionally, under unstimulated conditions, platelets from COVID-19 patients reported a slight increase in phosphatidyl serine exposure which was reduced upon stimulation. Further, the platelet-neutrophil interactions were observed to be increased in COVID-19 patients even under unstimulated conditions. P-selectin CD62P blocking antibody was observed to reduce understimulated basal platelet neutrophil interactions but had no impact on stimulated platelet-neutrophil interactions.
Therefore, the current study demonstrates the presence of two different platelet populations in COVID-19 patients. The first is circulating platelets with an altered proteome, and the second is P-selectin expressing neutrophil-associated platelets. The study also indicates that platelet-driven thromboinflammation is one of the critical factors that can increase the risk of thrombosis in COVID-19 patients. However, further research needs to be done to understand the mechanism of this effect.
Limitations
The study has certain limitations. First, heparin and dexamethasone may contribute to the platelet phenotype. Second, the healthy controls were younger with lower BMI. Third, the healthy controls self-reported being SARS-CoV-2 negative, which might be subjected to bias.
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
Goudswaard, L.J. et al. (2022). Alterations in platelet proteome signature and impaired platelet integrin αIIbβ3 activation in patients with COVID-19. bioRxiv. doi: https://doi.org/10.1101/2022.10.12.511145. https://www.biorxiv.org/content/10.1101/2022.10.12.511145v1
- Peer reviewed and published scientific report.
Goudswaard, Lucy J., Christopher M. Williams, Jawad Khalil, Kate L. Burley, Fergus Hamilton, David Arnold, Alice Milne, et al. 2023. “Alterations in Platelet Proteome Signature and Impaired Platelet Integrin ΑIIbβ3 Activation in Patients with COVID-19.” Journal of Thrombosis and Haemostasis, January. https://doi.org/10.1016/j.jtha.2023.01.018. https://www.jthjournal.org/article/S1538-7836(23)00059-4/fulltext.
 CDC tracks SARS-CoV-2 BA.3.2 global rise and finds early signals in U.S. wastewater
CDC tracks SARS-CoV-2 BA.3.2 global rise and finds early signals in U.S. wastewater