Molnupiravir, a widely utilized antiviral drug against SARS-CoV-2, induces changes in the viral genome at the time of replication. Although most random mutations will be harmful to the virus, some will be fatal.
In animal studies, higher mutation rates caused by molnupiravir have been shown to reduce viral burden. However, it is probable that some molnupiravir-treated patients might not completely clear SARS-CoV-2, thereby resulting in the transmission of viruses mutated by molnupiravir.
About the study
In the present study, researchers systematically explore global sequencing databases to identify a signature of molnupiravir-induced mutations.
The team analyzed a tree comprising more than 13 million SARS-CoV-2 sequences with annotated mutations obtained from the International Nucleotide Sequence Database Collaboration (INSDC) and Global Initiative on Sharing All Influenza Data (GISAID) databases. For every branch of the tree, the number of substitute classes was tallied. This tree was restricted to branches containing at least 20 substitutions, and the fraction of substitution types was plotted.
Noting that the signature also contained a high proportion of cytosine (C)-to-thymine (T) mutations, the team devised a criterion related to branches of interest. These were referred to as ‘high guanine (G)-to-adenine (A)’ branches, wherein branches with a minimum of 10 substitutions were selected, including a minimum of 25% G-to-A and 20% C-to-T substitutions, and no more than 5% transversions.
Next, the team determined whether the high G-to-A branches had a different mutation rate than other branches of comparable length. A date was assigned to every tree node.
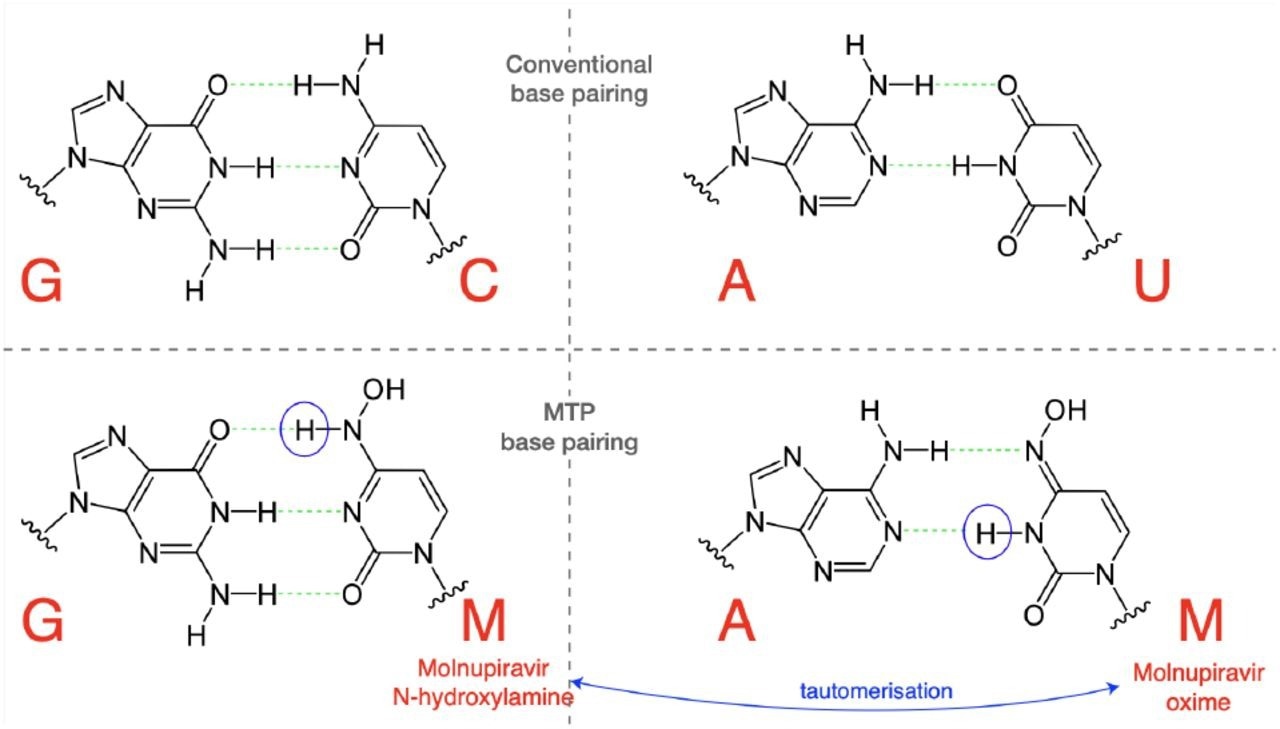 Molnupiravir triphosphate can assume multiple tautomeric forms. The N-hydroxylamine form resembles cytosine, while the oxime form more closely resembles uracil. They therefore pair with guanine and adenine respectively. (Figure adapted in part from Malone and Campbell (2021).)
Molnupiravir triphosphate can assume multiple tautomeric forms. The N-hydroxylamine form resembles cytosine, while the oxime form more closely resembles uracil. They therefore pair with guanine and adenine respectively. (Figure adapted in part from Malone and Campbell (2021).)
The mutational spectrum detected on these branches was then evaluated. The researchers reexamined the genomic dataset obtained from the AGILE Phase IIA clinical trial to compare the detected signals to mutations present in diagnosed molnupiravir-exposed patients. Furthermore, the range of suspected molnupiravir-induced alterations was evaluated by comparing day one and day five samples collected from the same patient.
Study findings
During the investigation of SARS-CoV-2 phylogenetic branches with numerous mutations, a subset with skewed proportions of mutation classes and minimal transversion substitutions were identified. These patterns differed significantly from the conventional SARS-CoV-2 mutational spectrum. An area of this space had a greater G-to-A ratio and only transition substitutions, thus indicating that a biological or technical change had led to a novel mutational signature.
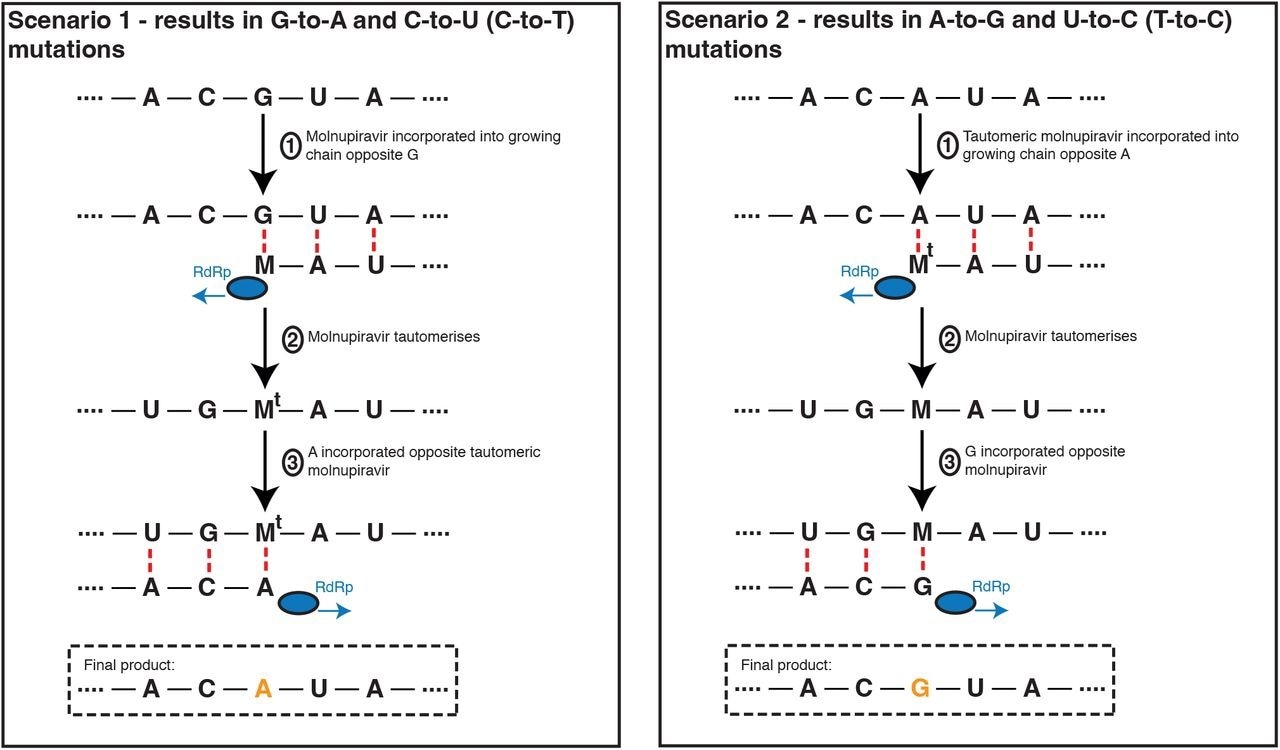 Molnupiravir drives G-to-A and C-to-U (C-to-T) mutations, and to a lesser extent A-to-G and T-to-U (T-to-C) mutations. In the most common scenario, shown on the left-hand side, M is incorporated opposite G nucleotides. It can then pair to A in subsequent replication, creating a G-to-A mutation. If the original G resulted from negative strand synthesis from a coding-strand C, then the G-to-A change ultimately creates a C-to-U coding change (Fig. S1). In the second, less common, scenario shown at the right hand side M is initially incorporated by pairing with A, which can result in an A-to-G mutation or, if the original A came from a coding U, a U-to-C mutation.
Molnupiravir drives G-to-A and C-to-U (C-to-T) mutations, and to a lesser extent A-to-G and T-to-U (T-to-C) mutations. In the most common scenario, shown on the left-hand side, M is incorporated opposite G nucleotides. It can then pair to A in subsequent replication, creating a G-to-A mutation. If the original G resulted from negative strand synthesis from a coding-strand C, then the G-to-A change ultimately creates a C-to-U coding change (Fig. S1). In the second, less common, scenario shown at the right hand side M is initially incorporated by pairing with A, which can result in an A-to-G mutation or, if the original A came from a coding U, a U-to-C mutation.
Numerous nations with many high G-to-A branches utilized molnupiravir. In the initial months of 2022, over 380,000 prescriptions were reported in Australia, over 30,000 in the United Kingdom, and over 240,000 in the United States. Age metadata obtained from the United States revealed a considerable bias toward older patients for the high G-to-A branches as compared to control branches with comparable numbers of mutations without any substitution type selection.
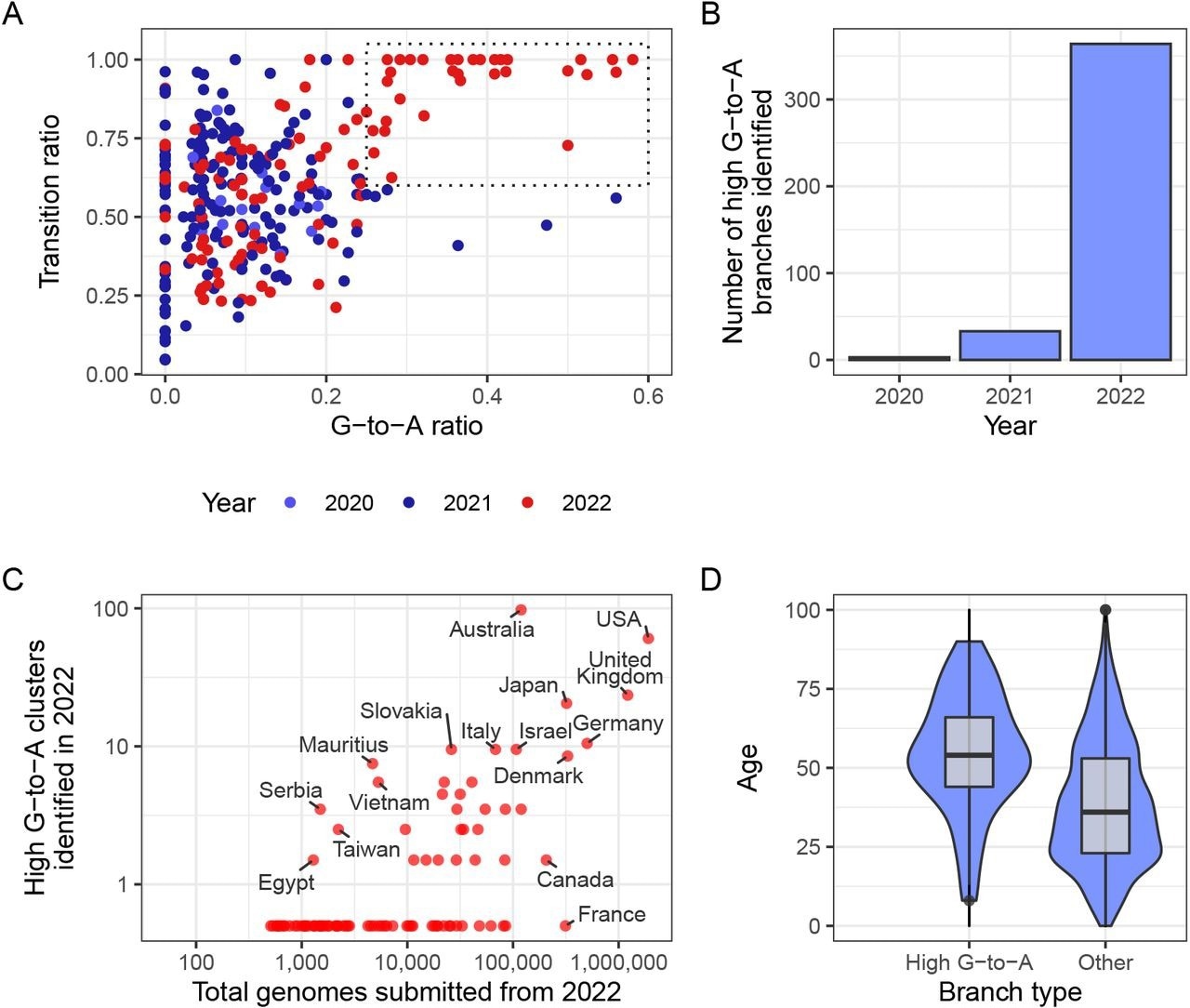 A new mutational signature emerged with high G-to-A and high transition ratio emerged in 2022 in some countries in global sequencing databases. (A) Each point in this scatterplot represents a branch from the mutation-annotated tree with >20 substitutions. Points are positioned according the proportion of the branch’s mutations that are G-to-A (x) or any transition mutation (y), and coloured by the year in which they occurred. A boxed region with higher G-to-A and almost exclusively transition mutations occurs only in 2022. (B) A count of the number of branches which satisfy a specific criterion (G-to-A ratio >= 25%, C-to-T ratio >= 20% transition ratio > 95%, total mutations > 10). (C) A comparison of number of total genomes with number of identified high G-to-A clusters (using the same criterion as B). Note logarithmic axes (semi-log, with the truncated line representing zero). For example, Australia has 97 clusters from a total of 119,194 genomes, whereas France has 0 clusters from 313,680 genomes. (D) A comparison of the age distributions for clusters with >10 mutations from the USA, partitioned by whether or not they satisfy the high G-to-A criterion. High G-to-A clusters correspond to older individuals.
A new mutational signature emerged with high G-to-A and high transition ratio emerged in 2022 in some countries in global sequencing databases. (A) Each point in this scatterplot represents a branch from the mutation-annotated tree with >20 substitutions. Points are positioned according the proportion of the branch’s mutations that are G-to-A (x) or any transition mutation (y), and coloured by the year in which they occurred. A boxed region with higher G-to-A and almost exclusively transition mutations occurs only in 2022. (B) A count of the number of branches which satisfy a specific criterion (G-to-A ratio >= 25%, C-to-T ratio >= 20% transition ratio > 95%, total mutations > 10). (C) A comparison of number of total genomes with number of identified high G-to-A clusters (using the same criterion as B). Note logarithmic axes (semi-log, with the truncated line representing zero). For example, Australia has 97 clusters from a total of 119,194 genomes, whereas France has 0 clusters from 313,680 genomes. (D) A comparison of the age distributions for clusters with >10 mutations from the USA, partitioned by whether or not they satisfy the high G-to-A criterion. High G-to-A clusters correspond to older individuals.
High G-to-A branches exhibited a distinct branch length distribution than other branch types, with an enrichment noted for branches with longer lengths. Furthermore, the branch length measured over time was shorter with respect to branches with a high G-to-A signature as compared to those with a matched length that lacked this signature, thereby indicating a higher mutation rate.
The identified spectrum was dominated by C-to-T and G-to-A transition mutations, while lesser contributions were noted from T-to-C and A-to-G transition mutations. This trend was consistent with molnupiravir's mechanisms of action.
These transitions displayed a propensity for specific nucleotide environments surrounding them. This may indicate a preference for the binding of molnupiravir adjacent to specific surrounding nucleotides, SARS-CoV-2 ribonucleic acid dependent RNA polymerase (RdRp) to add molnupiravir adjacent to particular nucleotides, or the viral proofreading exonuclease to eliminate molnupiravir in particular contextual environments.
The study also showed that molnupiravir-treated patients had considerably more viral mutations than placebo-treated patients. The spectrum of mutations between molnupiravir and placebo was significantly different.
While the patterns in transition were extremely similar, the AGILE trial molnupiravir spectrum comprised a much greater G-to-T mutational rate than the lengthy phylogenetic branches. This high rate was also noted in placebo-treated individuals, although it was comparatively higher in molnupiravir-treated persons.
Conclusions
The current study demonstrated that molnupiravir treatment signatures could be detected in global sequencing databases. More specifically, several long phylogenetic branches with an abundance of transition mutations were identified.
The findings also implied that, in at least some instances, viruses with a high number of molnupiravir-induced mutations have been transmitted to other persons, albeit in a restricted manner.
 *Important notice: medRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
*Important notice: medRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
 Study links COVID-19 to higher sleep apnea risk years after infection
Study links COVID-19 to higher sleep apnea risk years after infection