In a recent study published in the Genes journal, researchers assessed the impact of PARK2 duplication or microdeletion on neurological diseases such as Parkinson's.
The Parkinson's disease 2 (PARK2) gene encodes a protein with ubiquitin-E3-ligase activity, which has been identified as a p53 transcriptional repressor. The primary function of the Parkin protein is to control programmed cell death and mitophagy. This protein is expressed in various nervous system regions, including the basal ganglia, cerebellum, and cerebral cortex. PARK2 has been linked to various neurodevelopmental disorders (NDD), such as early-onset Parkinson's disease (PD), schizophrenia, attention-deficit/hyperactivity disorder (ADHD), and autism spectrum disorder (ASD). Several PARK abnormalities, including point mutations, triplications, duplications, copy number variants (CNVs) of varying sizes, and exonic deletions, have been implicated in NDD.
 Study: PARK2 Microdeletion or Duplications Have Been Implicated in Different Neurological Disorders Including Early Onset Parkinson Disease. Image Credit: MattLphotography / Shutterstock
Study: PARK2 Microdeletion or Duplications Have Been Implicated in Different Neurological Disorders Including Early Onset Parkinson Disease. Image Credit: MattLphotography / Shutterstock
About the study
In the present study, researchers at the University of Rochester Medical Center in the United States presented a series of patients with duplication/deletion at the 6q26 locus.
From 2008 to 2011, all patients with deletions/duplications at the 6q26 gene encompassing PARK2 were enrolled for the study from the aCGH laboratory dataset. If available, the patient's medical records were reviewed, or the majority of clinical data were extracted from the laboratory request form supplied with samples. Deoxyribonucleic acid (DNA) was extracted to conduct the array-based comparative genomic hybridization (aCGH) analysis, while fluorescence in situ hybridization (FISH) probes were acquired. Image acquisition and analysis were performed using applied spectral imaging (ASI) software. To confirm each aCGH discovery, 100 interphase, and 10 metaphase cells were investigated.
Results
The team discovered approximately nine patients in the study database whose aCGH tests revealed aberrations in the PARK2 gene copy number. The first patient was a nine-year-old girl with a history of seizures, developmental delay, encephalopathy, and dysmorphic features. Array CGH assessment revealed that the first patient had duplications of 506 Kb and 347 kb at chromosome 17q21.3–17q21.32 and chromosome 6q26 region containing the PARK2 gene, respectively. The chromosome 17q21.3–17q21.32 aberration affects the genes adenosine diphosphate (ADP)-ribosylation factor-like protein 17A (ARL17A), N-ethylmaleimide-sensitive factor (NSF), leucine-rich repeat-containing 37A (LRRC37A), ARL17P1, K(lysine) acetyltransferase-8 regulatory nonspecific-lethal complex subunit 1 (KIAA1267), Homo sapiens ADP-ribosylation factor-like protein variant (LOC51326), and Homo sapiens hypothetical protein (LOC644246). FISH studies of parental samples revealed paternal inheritance.
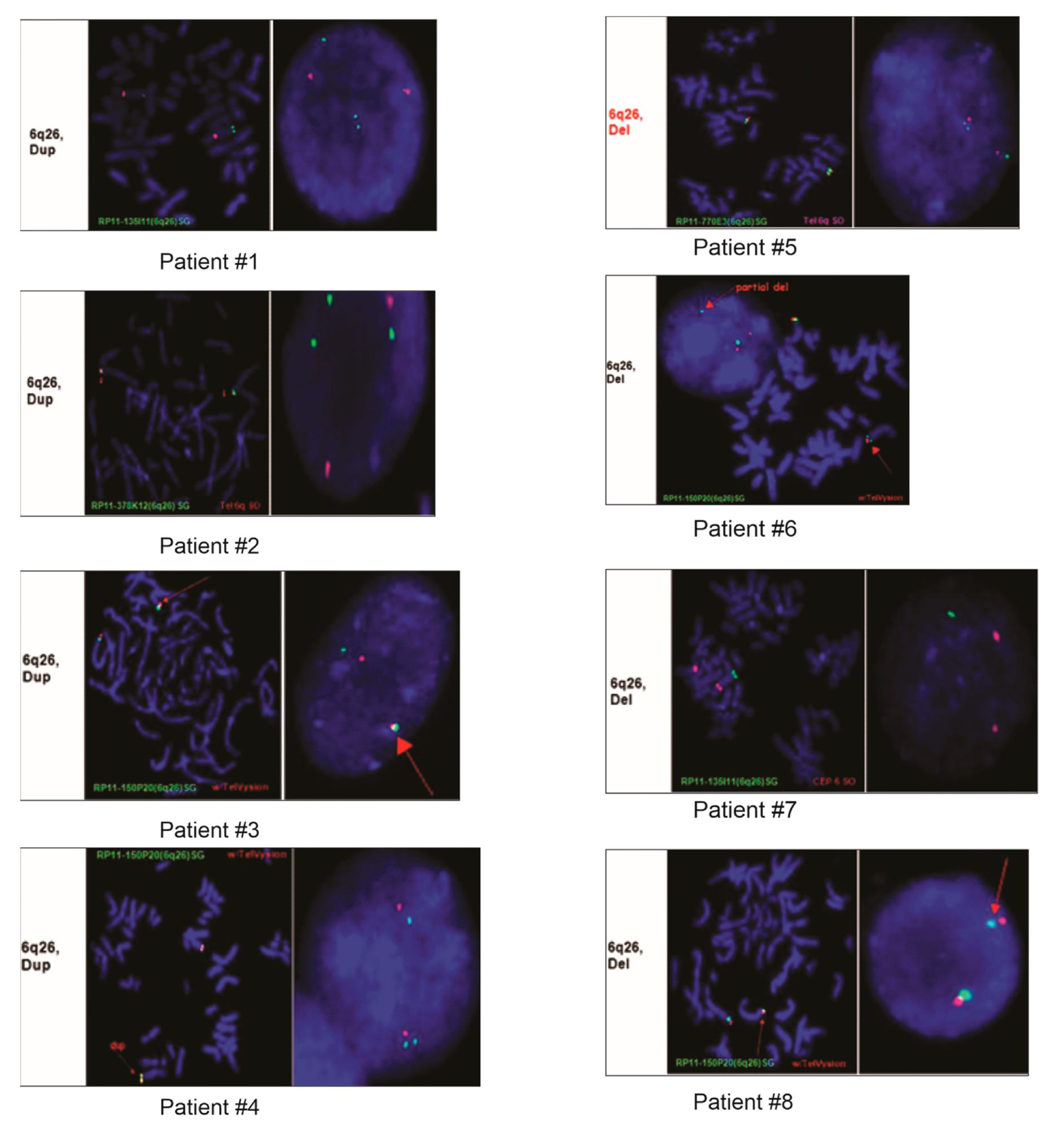 FISH images from eight of the nine patients demonstrating duplication or deletion within the 6q26 region.
FISH images from eight of the nine patients demonstrating duplication or deletion within the 6q26 region.
The second patient was a one-year-old boy referred for Dandy-Walker disease and hypotonia. He displayed a duplication of 726 Kb on the chromosome 6q26 region containing the PACRG and PARK2 genes, as revealed by array CGH analysis. FISH studies of parental samples noted maternal inheritance. The third patient was a one-day-old fetus that died immediately after birth. The autopsy revealed that placenta abruption was the cause of death. Array CGH analysis of fetal tissue revealed a 279 kb duplication at the chromosome 6q26 gene.
The fourth patient was a 28-year-old woman with a history of seizures and developmental delays. Array CGH performed on the material submitted revealed a duplication of 476 kb at the chromosome 6q26 gene. The fifth patient was a five-year-old girl referred for assessment because of a congenital abnormality and had a duplication of 120 kb on chromosome 20p13 and a deletion of 215 kb on chromosome 6q26 gene. The genes β-defensin 126 (DEFB126), DEFB125, DEFB128, and DEFB127 were affected by chromosome 20p13 aberration. FISH studies of parental samples demonstrated de novo origin. The sixth patient was a nine-year-old boy who underwent an aCGH testing for autism and developmental deficits. The aCGH analysis revealed 346 kb deletion at the chromosome 6q26 gene.
The seventh patient was a two-year-old boy who underwent an aCGH due to a history of developmental difficulties. Array CGH results revealed copy-number alterations (CNAs) on chromosome 17p13.3, chromosome 19p12, and chromosome 7q35, as well as a 252 kb deletion on chromosome 6q26. The eighth patient was a two-month-old girl who had a history of dysmorphic traits and hypotonia. Array CGH indicated a 216 kb deletion on the chromosome 6q26 locus, and FISH analysis of parental samples revealed that the deletion was paternally inherited. Lastly, the ninth patient was a symptom-free 39-year-old man who underwent array CGH testing due to a family history of microdeletion at the 6q26 gene. The array CGH results of this patient indicated a chromosome 6q26 deletion of 216 kb.
Conclusion
The study findings showed nine novel examples of microdeletion/microduplication involving exons seven to 10 of PARK2. Other CNVs identified in three of the assessed patients have been associated with a wide range of neurodevelopmental disorders with 17q21.31–q21.32 duplication in the first patient, 20p13 duplication in the fifth patient, and 7q35 deletion and 17p13.3 and 19p12 duplication in the seventh patient. Whether these extra CNVs promote early illness manifestation or severe disease in PARK2 patients is still uncertain. However, the researchers believe that additional CNVs are more likely to influence the diverse clinical phenotypes exhibited by these patients.
 Fish-inspired device enables scalable monitoring of beating heart organoids
Fish-inspired device enables scalable monitoring of beating heart organoids