New genome study uncovers 133 previously unknown dementia-linked variants and shows how genetic risks vary across global populations, reshaping the path to personalized Alzheimer’s care.
 Study: Biobank-scale genetic characterization of Alzheimer’s disease and related dementias across diverse ancestries. Image Credit: BlueBackIMAGE / Shutterstock
Study: Biobank-scale genetic characterization of Alzheimer’s disease and related dementias across diverse ancestries. Image Credit: BlueBackIMAGE / Shutterstock
In a recent paper in the journal Nature Communications, researchers examined genetic factors associated with Alzheimer’s disease and other dementias across 11 ancestral groups. Their aim was to identify which gene variants promote resilience and protection, and which may confer risk or act as potential disease-causing variants.
They found differences in how genetic variants, particularly in the apolipoprotein E (APOE) gene, influence the risk of developing dementia across ancestries. The APOE gene is critical for lipid metabolism. Its ε4 variant is significantly associated with the risk of developing Alzheimer’s disease, though its impact is modulated by ancestry and additional genetic modifiers, including a protective locus at 19q13.31 observed in African ancestry populations.
Background
Dementia currently affects around 55 million people worldwide and is projected to nearly triple by 2050, with Alzheimer’s disease being the most common form. Despite advances in genetics, most research has focused on European ancestry populations, limiting applicability to global populations.
Since genetic risk factors vary across ancestries, including diverse groups is critical for developing precise and equitable therapies. Large-scale global datasets now offer opportunities to study how genetic, environmental, and clinical factors influence dementia. However, dementia-specific datasets remain limited.
A major gap lies in understanding protective and resilience variants, genetic factors that delay onset, lessen severity, or reduce disease risk, often through modifying known risk variants such as APOE ε4. Only a handful of such variants have been identified to date.
About the Study
Researchers addressed these gaps by examining multi-ancestry whole-genome sequencing data, aiming to uncover genetic causes, risks, and protective mechanisms in Alzheimer’s disease and related dementias, with implications for personalized therapeutics worldwide.
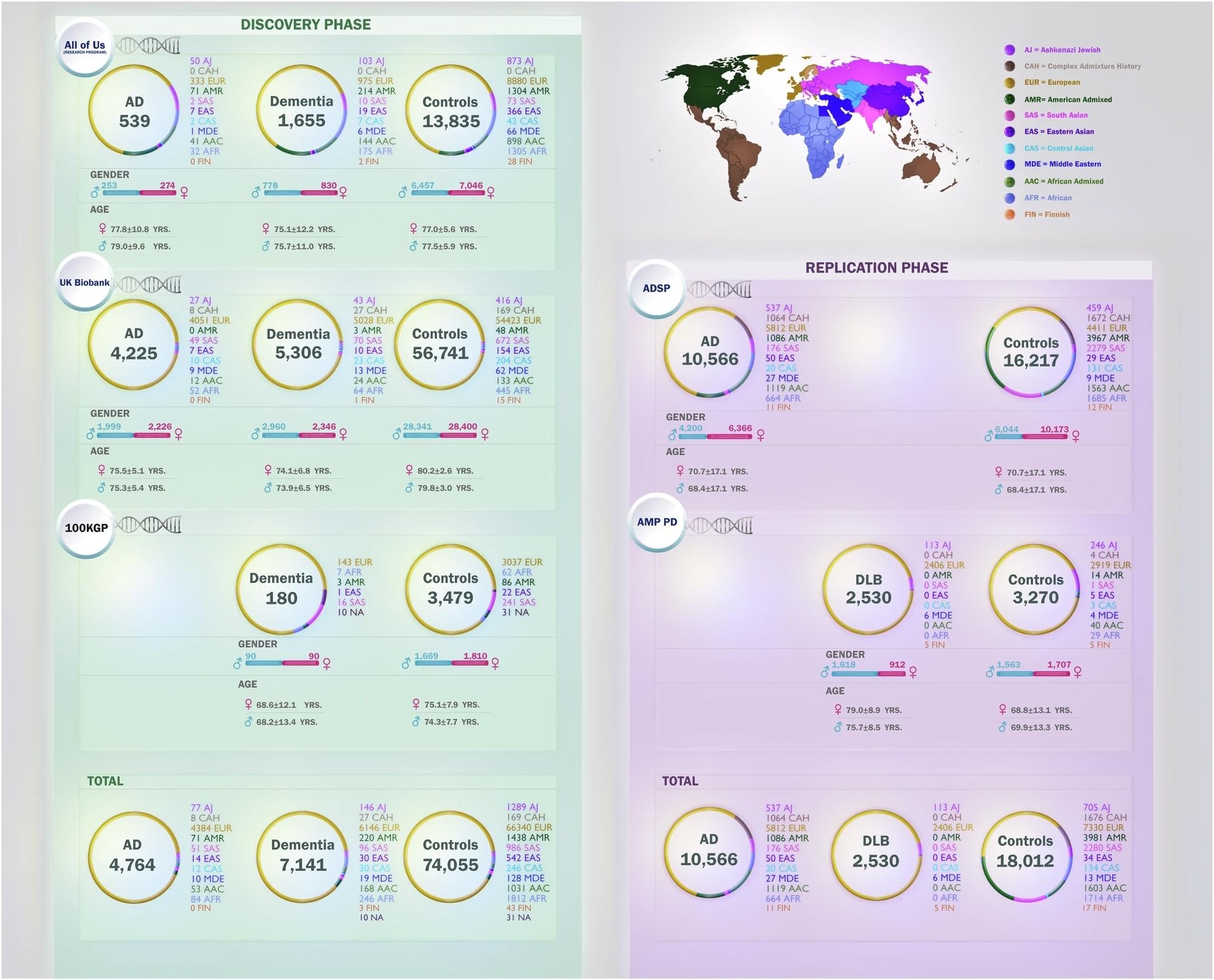 The figure illustrates distributions of age, sex, and the number of cases and controls per ancestry across five datasets in this study: All of Us (AoU), Alzheimer’s Disease Sequencing Project (ADSP), 100,000 Genomes Project (100KGP), UK Biobank (UKB), and Accelerating Medicines Partnership in Parkinson’s Disease (AMP PD). Ancestries represented include European (EUR), African (AFR), American Admixed (AMR), African Admixed (AAC), Ashkenazi Jewish (AJ), Central Asian (CAS), Eastern Asian (EAS), South Asian (SAS), Middle Eastern (MDE), Finnish (FIN), and Complex Admixture History (CAH).
The figure illustrates distributions of age, sex, and the number of cases and controls per ancestry across five datasets in this study: All of Us (AoU), Alzheimer’s Disease Sequencing Project (ADSP), 100,000 Genomes Project (100KGP), UK Biobank (UKB), and Accelerating Medicines Partnership in Parkinson’s Disease (AMP PD). Ancestries represented include European (EUR), African (AFR), American Admixed (AMR), African Admixed (AAC), Ashkenazi Jewish (AJ), Central Asian (CAS), Eastern Asian (EAS), South Asian (SAS), Middle Eastern (MDE), Finnish (FIN), and Complex Admixture History (CAH).
They combined large-scale datasets to identify and evaluate genetic variants linked to Alzheimer’s disease and related dementias, from biobanks across the United States, the United Kingdom, the 100,000 Genomes Project, Alzheimer’s Disease Sequencing Project, and Accelerating Medicines Partnership for Parkinson’s disease.
In the discovery phase, the first three datasets were used. Cases were identified through electronic health records, clinical assessments, or registry-linked diagnoses, while controls were defined as individuals aged 65 and above without neurological conditions.
Whole-genome sequencing was performed under standardized protocols, and data underwent strict quality control to exclude low-quality samples, duplicates, and closely related individuals. They filtered variants to focus on protein-altering and splicing mutations, calculating allele frequencies and zygosity across ancestries.
The replication phase used the Alzheimer’s Disease Sequencing Project and Accelerating Medicines Partnership for Parkinson’s disease datasets, applying similar quality control measures. Variants prioritized included those classified as pathogenic, risk, or protective, based on established clinical and population databases.
Analyses also examined APOE genotypes and their modifiers, rare variant burdens, and polygenic risk scores. Ancestry was assigned using reference datasets and computational pipelines to ensure population-level comparisons.
Key Findings
This large-scale genetic study analyzed three major datasets to identify 156 variants, including 133 novel variants, linked to Alzheimer’s disease and related dementias. Across these cohorts, researchers detected hundreds of variants in genes already associated with neurodegenerative diseases, including APOE.
Many variants were rare, heterozygous, and predicted to be potentially deleterious. Several known mutations confirmed previous links to Alzheimer’s disease, frontotemporal lobar degeneration (FTLD), or other disorders, while many novel variants were identified in underrepresented ancestries such as African, South Asian, and Ashkenazi Jewish populations.
The study also identified ancestry-specific modifiers of APOE ε4 risk, with evidence of protective effects observed in certain loci within African populations, with implications for precision medicine.
Replication analyses in independent datasets validated a subset of both known and novel variants across different ancestries. Some variants were found only in cases, while others also appeared in controls, suggesting incomplete penetrance, possible misclassification, or potential roles as risk factors rather than direct causes.
Importantly, the study highlighted APOE ε4’s strong but variable impact across ancestries, with additional protective or disease-modifying variants influencing risk in some populations. Burden analysis revealed enrichment of rare pathogenic variants in the glucosylceramidase beta 1 (GBA1) gene. Overall, the findings expand knowledge of both established and novel genetic contributors to multiple dementias across diverse populations.
Conclusions
This study provides a comprehensive catalog of coding and splicing variants in known genes associated with dementia risk, including 133 novel variants across diverse populations. Replication across datasets strengthens their potential pathogenicity, though functional validation remains essential.
Notably, many variants were ancestry-specific, with 26 potentially disease-causing variants identified in non-European groups, 18 of which were absent in Europeans, underscoring the need for inclusive genetics research. Some variants previously labeled pathogenic were also present in controls, highlighting challenges of incomplete penetrance, potential misdiagnosis, and the importance of careful interpretation.
Genotype–phenotype analyses reinforced known associations while suggesting novel variants linked to early-onset dementia. However, limitations include underrepresentation of certain ancestries, reliance on heterogeneous datasets, lack of neuropathological confirmation, and challenges in detecting repeat expansions.
Despite these, the findings and the publicly available MAMBARD browser offer valuable resources for global research in dementia and the development of effective therapies.
Journal reference:
- Biobank-scale genetic characterization of Alzheimer’s disease and related dementias across diverse ancestries. Khani, M., Akçimen, F., Grant, S.M., Akerman, S.C., Lee, P.S., Faghri, F., Leonard, H., Kim, J.J., Makarious, M.B., Koretsky, M.J., Rothstein, J.D., Blauwendraat, C., Nalls, M.A., Singleton, A., Bandres-Ciga, S. Nature Communications (2025). DOI: 10.1038/s41467-025-62108-y, https://www.nature.com/articles/s41467-025-62108-y