Around 10% of the global population is affected by chronic kidney disease (CKD). The risk of CKD progressing into end-stage renal disease (ESRD) is exceptionally high, which requires dialysis or kidney transplantation. At present, there is no effective treatment for CKD is available. Hence, there is an urgent need to uncover the underlying pathological mechanisms of CKD to help formulate effective treatment strategies to prevent and cure the disease. A recent Nature Communications study suggested that DNA-PKcs could be a potential target for treating CKD.
An important pathological characteristic of CKD is renal interstitial fibrosis, associated with an unusual expression of profibrotic factors, such as transforming growth factor-beta 1 (TGF-β1), myofibroblast activation, and epithelial de-differentiation. TGF-β1 has a significant role in interstitial fibrosis that includes activation of fibrotic genes, such as fibronectin (FN), α-smooth muscle actin (α-SMA), and collagens. In addition, it is also associated with the Warburg effect-like metabolic reprogramming of kidney cells.
While analyzing the association between metabolic dysregulation and interstitial fibrosis, researchers observed metabolic reprogramming of kidney cells, i.e., myofibroblasts and renal tubular epithelial cells, during kidney injury. This occurrence influences the development of CKD.
During the metabolic reprogramming of kidney cells, a significant reduction in fatty acid oxidation (FAO) occurs along with a metabolic shift to glycolysis. These manifestations result in immune cell infiltration and interstitial fibrosis. Several animal models of renal fibrosis have shown that fibrosis attenuation is possible through glycolysis inhibition and FAO restoration using genetic or pharmacological approaches.
DNA-dependent protein kinase (DNA-PK), which is a trimeric complex consisting of a catalytic subunit (DNA-PKcs) and a Ku70/80 heterodimer, is activated by reactive oxygen species (ROS) or DNA double-stranded breaks (DSBs). DNA-PK facilitates nonhomologous end joining (NHEJ) by connecting programmed DSBs, which is extremely important for lymphocyte recombination. Therefore, DNA-PKcs mutations inhibit the development of T and B lymphocytes.
DNA-PKcs plays an essential role in various metabolic functions, such as phosphorylation of the transcription factor USF-1 that promotes fatty acid synthesis induced by insulin and metabolic deterioration during aging. In addition, a previous study revealed that DNA-PKcs regulates the target of rapamycin (mTOR) activation. Even though DNA damage response (DDR) has been linked to renal epithelial injury, little evidence has been documented about its role in epithelial de-differentiation and myofibroblast activation in progressive CKD.
About the Study
The expression of DNA-PKcs was found to be increased in fibrotic kidneys, which progresses CKD development. The current study also revealed that DNA-PKcs expression was induced by TGFβ1-SMAD signaling, and DNA-PKcs knockout blocked SMAD2/SMAD3. These findings indicate a new pathway associated with TGFβ1-SMAD signaling activation in fibrosis.
A global prkdc gene-knockout mice model was developed and was used in this study. In vivo experiments using this mice model suggested that the elimination of DNA-PKcs resulted in attenuated renal tubular injury along with a reduction in the progression of renal interstitial fibrosis in unilateral ureteral obstruction (UUO) and unilateral ischemia-reperfusion (UIR) mouse models.
The current study failed to confirm whether DNA-PK promotes renal fibrosis independent of lymphocytes because lymphocyte deficiency is the most prominent phenotype of DNA-PKcs−/− mice. Therefore, to remove the effect of lymphocyte deficiency, the authors generated proximal renal tubular epithelial cells possessing specific DNA-PKcs knockout in vivo, using CRISPR/cas9 knock-in mice.
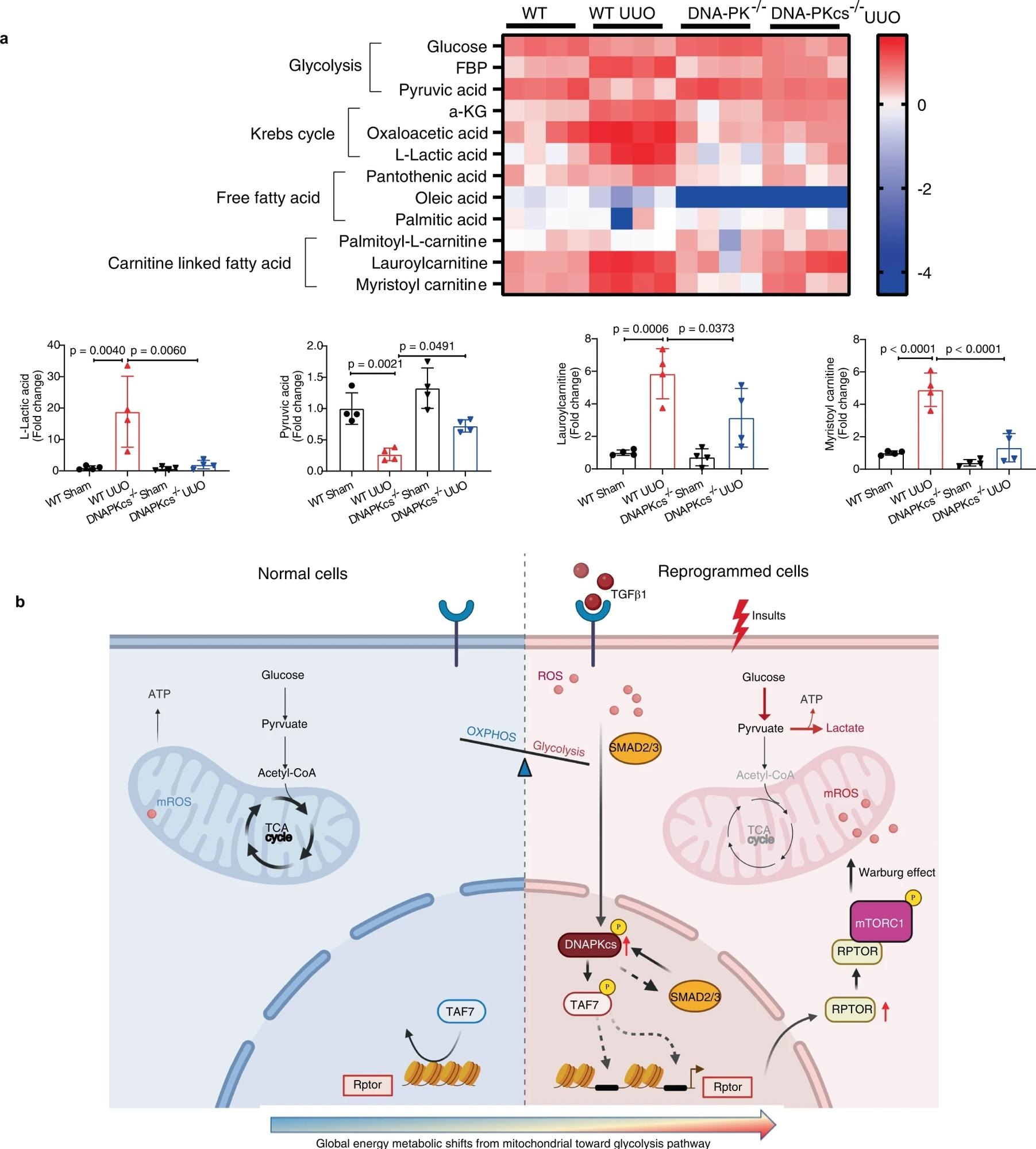 a Metabolomics analysis of kidney tissues from each group as indicated. Heatmap image showing relative levels of metabolites in the glycolysis pathway, fatty acid metabolism and Krebs cycle in kidneys from each group (n = 4). Bars represent statistical analysis of representative metabolites in kidneys of each group (mean ± SD, n = 4 mice of each group). One-way ANOVA followed by Tukey’s multiple comparisons test was used to determine the p-values. b Working model (the template was created with BioRender.com) illustrating in which DNA-PKcs mediates activation of Raptor/mTORC1 signaling through phosphorylation of TAF7 and promotes metabolic reprogramming in injured epithelial cells and myofibroblasts.
a Metabolomics analysis of kidney tissues from each group as indicated. Heatmap image showing relative levels of metabolites in the glycolysis pathway, fatty acid metabolism and Krebs cycle in kidneys from each group (n = 4). Bars represent statistical analysis of representative metabolites in kidneys of each group (mean ± SD, n = 4 mice of each group). One-way ANOVA followed by Tukey’s multiple comparisons test was used to determine the p-values. b Working model (the template was created with BioRender.com) illustrating in which DNA-PKcs mediates activation of Raptor/mTORC1 signaling through phosphorylation of TAF7 and promotes metabolic reprogramming in injured epithelial cells and myofibroblasts.
Notably, renal tubular-specific deletion of DNA-PKcs was also found to hamper the advancements of renal interstitial fibrosis in UUO. In vitro experiments revealed that DNA-PKcs deficiency was able to preserve the tubular epithelial cell phenotype and regulate interstitial fibroblast activation in vitro. These findings suggest that DNA-PKcs facilitate myofibroblast activation and epithelial de-differentiation without any direct link to lymphocyte deficiency.
The antifibrotic effects of NU7441, a highly specific DNA-PKcs inhibitor, were studied. In both UUO and UIR mouse models, NU7441 treatment was able to significantly attenuate the progression of renal interstitials. At physiological doses, NU7441 could partially inhibit DNA-PK.
Both in vivo and in vitro studies showed that DNA-PKcs deficiency did not exacerbate DSBs, indicating DNA-PKcs mediates renal injury and renal interstitial fibrosis. Furthermore, Phosphoproteomics analysis revealed a decreased phosphorylation of TAF7 in the kidney tissues of DNA-PKcs−/− mice. Interestingly, TAF7 deficiency blocked the profibrotic phenotype of fibroblasts and renal epithelial cells triggered by TGFβ1.
The profibrotic effects of TAF7 were almost entirely blocked by DNA-PKcs deficiency in renal epithelial cells. Our results indicate that TAF7 is a substrate for DNA-PKcs kinase activity and that DNA-PKcs-mediated phosphorylation of TAF7 aggravates renal fibrosis. ChIP assay showed that TAF7 can bind to the Rptor promoter directly, however, the underlying mechanism must be elucidated in future research. The authors pointed out that DNA-PKcs mediates the activation of RAPTOR/mTORC1 signaling through phosphorylation of TAF7.
Conclusions
DNA-PKcs activity was found to be undetectable in normal kidneys, in contrast to a significant increase during CKD. DNA-PKcs facilitates activation of RAPTOR/mTORC1 signaling via phosphorylation of TATA-box binding protein associated factor 7 (TAF7) in CKD. DNA-PKcs inhibition restores metabolic reprogramming in injured kidney cells, such as epithelial cells and myofibroblasts, in CKD. Hence, DNA-PKcs could be an important target for CKD treatment.
-590_18da4e909d704b5fa71b7dd9ad43e610-80x70.jpg) Novel DNA nanoplatform offers hope for chronic gum disease
Novel DNA nanoplatform offers hope for chronic gum disease