 Study: Interstitial macrophages are a focus of viral takeover and inflammation in COVID-19 initiation in human lung. Image Credit: Dotted Yeti / Shutterstock
Study: Interstitial macrophages are a focus of viral takeover and inflammation in COVID-19 initiation in human lung. Image Credit: Dotted Yeti / Shutterstock
About the study
In the present study, researchers developed an experimental COVID-19 model to investigate early molecular processes and pathogenic mechanisms of SARS-CoV-2 infection at the cellular level in native tissues of the human lung.
The researchers established SARS-CoV-2's cellular tropism and its unique and dynamic impacts on host cellular gene expression in specific types of lung cells. Prominent targets were lung-resident macrophages, of which one SARS-CoV-2 takes over transcriptomes, inducing a targeted host interferon (IFN) antiviral program, and several chemokines and pro-fibrotic and pro-inflammatory and cytokines signaling to various structural and immunological cells of the lung.
To determine the early stages of COVID-19 in human lungs, the researchers sliced lung tissue obtained from surgical specimens or organ donor individuals into thick sections and used them for tissue culture analysis. Subsequently, they exposed the tissues to the SARS-CoV-2 USA-WA1 2020 strain at 1.0 multiplicity of infection (MOI) for two hours before allowing the SARS-CoV-2 infection to continue for two to three days. They performed a plaque test on culture supernatants.
The researchers separated the slices and examined them by scRNA-seq to evaluate host and viral genetic expression during the SARS-CoV-2 infection. They also examined the viral RNA molecules' junctional structure and processing by analyzing the scRNA-seq dataset with the SICILIAN framework. They used molecular atlas markers to distinguish lung cell types in healthy lung slices and measure viral RNA levels in infected cells.
The team performed multiplexed single-molecule fluorescence in situ hybridization (smFISH) to confirm lung cell tropism findings and show infected cells. They used single-cell gene expression patterns to identify cellular targets for inflammatory and pro-fibrotic signals elicited by the SARS-CoV-2 infection of a-IMs. They devised a technique for purifying macrophage populations from human lungs with a SARS-CoV-2 spike (S) protein-pseudotyped lentivirus (lenti-S-NLuc-tdT) to investigate lung macrophage entrance routes.
The researchers productively infected human lung slices cultivated ex vivo with SARS-CoV-2, with production rising between 24 and 72 hours of culture. They heat-inactivated, ultraviolet (UV)-treated, or administered 10.0 µM remdesivir, an RNA-dependent RNA polymerase inhibitor used as a COVID-19 therapeutic, to prevent viral stock infection.
Results
The analysis showed that SARS-CoV-2 preferentially infects active interstitial macrophages (IMs), which can amass hundreds of SARS-CoV-2 RNA molecules, comprising >60% of the cell transcriptome and producing dense viral RNA bodies. Infected alveolar macrophages (AMs) exhibit no severe reactions, with spike (S) protein-dependent viral entrance into AMs utilizing angiotensin-converting enzyme 2 (ACE2) and the cluster of differentiation 169 (CD169) and IM entry via CD209.
They found canonical sub-genomic junctions between the unusual sequence reads beyond their 39 terminal regions, indicating canonical-type SARS-CoV-2 messenger RNA (mRNA) production in the pulmonary cultures. They also found hundreds of new subgenomic junctions, showing a wide range of non-canonical and canonical sub-genomic SARS-CoV-2 RNAs produced during pulmonary infection.
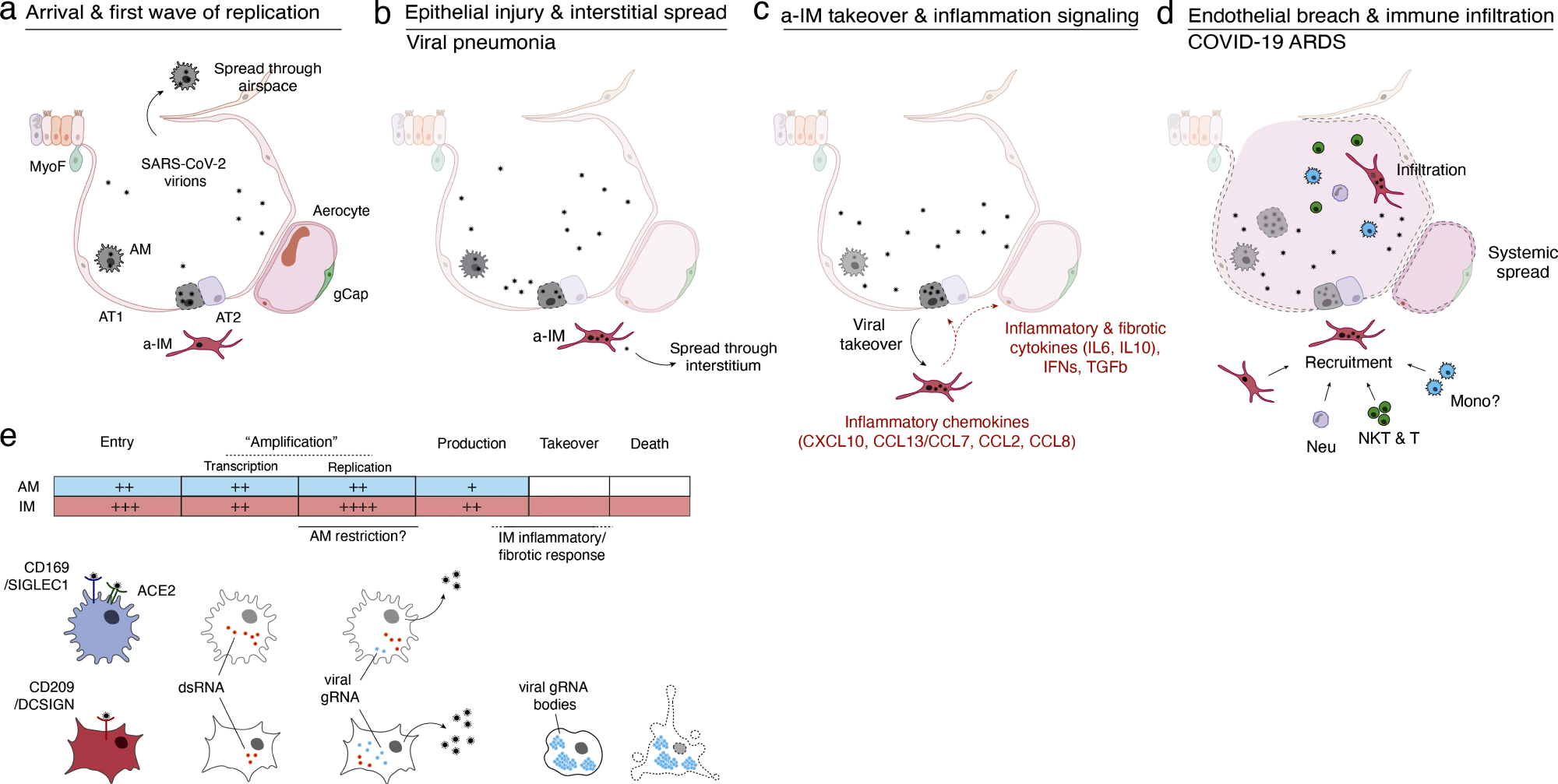 Model of initiation, transition, and pathogenesis of COVID-19 and the viral lifecycle in AMs and IMs. (a–d) Model of COVID-19 initiation in the human lung and transition from viral pneumonia to lethal COVID-19 ARDS. (a) SARS-CoV-2 virion dissemination and arrival in the alveoli. Luminal AM encounter virions shed from the upper respiratory tract that enter the lung. AMs can express low to moderate numbers of viral RNA molecules and can propagate the infection but “contain” the viral RNA from taking over the total transcriptome and show only a very limited host cell inflammatory response to viral infection. (b) Replication and epithelial injury. SARS-CoV-2 virions enter AT2 cells through ACE2, its canonical receptor, and “replicate” to high viral RNA levels, producing infectious virions and initiating viral pneumonia. (c) a-IM takeover and inflammation signaling. SARS-CoV-2 virions spread to the interstitial space through either transepithelial release of virions by AT2 cells or injury of the epithelial barrier, and enter a-IMs. Infected a-IMs can express very high levels of viral RNA that dominate (“take over”) the host transcriptome and can propagate the infection. Viral takeover triggers induction of the chemokines and cytokines shown, forming a focus of inflammatory and fibrotic signaling. (d) Endothelial breach and immune infiltration. The a-IM inflammatory cytokine IL6 targets structural cells of the alveolus causing epithelial and endothelial breakdown, and the inflammatory cytokines recruit the indicated immune cells from the interstitium or bloodstream, which flood and infiltrate the alveolus causing COVID-19 ARDS. Local inflammatory molecules are amplified by circulating immune cells, and reciprocally can spread through the bloodstream to cause systemic symptoms of cytokine storm. (e) Comparison of the SARS-CoV-2 viral lifecycle in AMs and IMs. Although both can produce infectious virions, note differences in viral entry receptors (AMs can use ACE2 and CD169/SIGLEC1, whereas IMs use CD209); viral RNA transcription of dsRNA intermediates (greater in AMs); replication of full-length genomic RNA (greater in IMs); viral takeover, formation of RNA bodies, and induction of a robust host cell inflammatory response (only in IMs), and cell destruction/death (only in IMs).
Model of initiation, transition, and pathogenesis of COVID-19 and the viral lifecycle in AMs and IMs. (a–d) Model of COVID-19 initiation in the human lung and transition from viral pneumonia to lethal COVID-19 ARDS. (a) SARS-CoV-2 virion dissemination and arrival in the alveoli. Luminal AM encounter virions shed from the upper respiratory tract that enter the lung. AMs can express low to moderate numbers of viral RNA molecules and can propagate the infection but “contain” the viral RNA from taking over the total transcriptome and show only a very limited host cell inflammatory response to viral infection. (b) Replication and epithelial injury. SARS-CoV-2 virions enter AT2 cells through ACE2, its canonical receptor, and “replicate” to high viral RNA levels, producing infectious virions and initiating viral pneumonia. (c) a-IM takeover and inflammation signaling. SARS-CoV-2 virions spread to the interstitial space through either transepithelial release of virions by AT2 cells or injury of the epithelial barrier, and enter a-IMs. Infected a-IMs can express very high levels of viral RNA that dominate (“take over”) the host transcriptome and can propagate the infection. Viral takeover triggers induction of the chemokines and cytokines shown, forming a focus of inflammatory and fibrotic signaling. (d) Endothelial breach and immune infiltration. The a-IM inflammatory cytokine IL6 targets structural cells of the alveolus causing epithelial and endothelial breakdown, and the inflammatory cytokines recruit the indicated immune cells from the interstitium or bloodstream, which flood and infiltrate the alveolus causing COVID-19 ARDS. Local inflammatory molecules are amplified by circulating immune cells, and reciprocally can spread through the bloodstream to cause systemic symptoms of cytokine storm. (e) Comparison of the SARS-CoV-2 viral lifecycle in AMs and IMs. Although both can produce infectious virions, note differences in viral entry receptors (AMs can use ACE2 and CD169/SIGLEC1, whereas IMs use CD209); viral RNA transcription of dsRNA intermediates (greater in AMs); replication of full-length genomic RNA (greater in IMs); viral takeover, formation of RNA bodies, and induction of a robust host cell inflammatory response (only in IMs), and cell destruction/death (only in IMs).
Heat, UV-C inactivation, or remdesivir therapy prevented the development of canonical and non-canonical connections. The team observed SARS-CoV-2 takeover of an activated IM subtype in 176,382 cells with high-quality transcriptomes obtained from infected lung slices of four donor lungs and in 112,359 cells from mock-infected slices (cultured without viral addition) and 95,389 uncultured control cells (directly from freshly cut lung slices). A differential gene expression study of a-IMs over infection pseudotime revealed host gene expression alterations corresponding to SARS-CoV-2 RNA levels.
The study found that COVID-19 pneumonia infection and takeover cause an early antiviral cell response specific to activated interstitial macrophages, resulting in a powerful immunological and fibrotic signaling center. Inflammasome activation is uncommon and only detectable late in a-IM infection. Blocking antibodies against CD169 and CD209 prevented entrance into IMs and AMs. The study also highlighted IMs as the most vulnerable lung target, with initial emphasis on inflammation and fibrosis. Two unique molecular lineages of macrophage targets react differently to SARS-CoV-2, influencing etiology and treatments.
 Study links COVID-19 to higher sleep apnea risk years after infection
Study links COVID-19 to higher sleep apnea risk years after infection