A revision of the EU GMP Volume 4 Annex 1 was issued in August 2022, introducing new regulations for sterile drug products and production. This release replaced the 2020 draft and 2008 version, adding emphasis to the importance of validation and qualification.
The new version of Annex 1 is much longer, consisting of 59 pages instead of the previous version’s 16 pages. This additional content provides greater detail on the specific needs and requirements for premises, utilities, equipment, and personnel, offering improved guidance for the manufacturing of sterile products.
Some principles and guidance outlined in the Annex may also be used to support the manufacturing of products that are not intended to be sterile, including contamination control strategies, premises design, cleanroom classification, validation, qualification, monitoring, and personnel gowning.
These non-sterile applications place great importance on controlling and reducing particulate, microbial, and endotoxin/pyrogen contamination.1
Beyond the Annex 1 document itself, Part 1, Chapter 3 of the Eudralex Volume 4 outlines the fundamental requirements and characteristics for premises and equipment to prevent cross-contamination, dirt or dust build-up, and other adverse effects on product quality.
However, in the initial section of the new Annex 1, facilities, equipment, and processes are included in the process life cycle.
It is important that products and processes are designed, qualified, validated, and, where applicable, subjected to ongoing verification in line with relevant sections of the Good Manufacturing Practices (GMP) guidelines.
Annex 1 considers this the first special requirement for minimizing the risks of particulate, microbial, and endotoxin/pyrogen contamination. Each pharmaceutical process, utility, system, or piece of equipment should be designed and constructed for a defined purpose, with the respective qualification or validation demonstrating adherence to that purpose.
Validation and qualification also play a key role in identifying a drug product’s potential critical quality attributes (CQAs). These core product characteristics affect product quality, meaning it is advisable to study and control them as part of a comprehensive control strategy.2
It is important to manage processes, facilities, equipment, and manufacturing activities in line with quality risk management (QRM) principles. This should be the case for all steps, including and following the design stage, and it is essential that manufacturers understand their processes and control their variability.
Understanding validation and qualification
The distinction between validation and qualification can be unclear. These terms are often used interchangeably, but it is often more appropriate to broaden the concept of ‘validation’ to include the concept of qualification.
GMP principles consider both validation and qualification as actions to demonstrate that expected results have been achieved, but there is a small difference between the two.
For example, validation ensures that any process, procedure, equipment, material, system, or activity leads to the anticipated results, while qualification is the act of demonstrating that equipment works correctly.
Validation is central to the pharmaceutical manufacturing industry, yet definitions vary across the field. One of these definitions is provided by Agalloco:
Validation is a defined program, which in combination with routine production methods and quality control techniques, provides documented assurance that a system is performing as intended and/or that a product conforms to its predetermined specifications. When practiced in a life-cycle model, it incorporates design, development, evaluation, operation and maintenance considerations to provide both operating benefits and regulatory compliance.3
Agalloco
This definition highlights key quality attributes that must be considered when thinking about validation, including consistency in terms of reproducibility and design (planned, pre-determined, and performed by trained personnel), and documented evidence (data integrity).
The concept of validation can be associated with processes, including the initial validation of new processes (e.g., regulatory submissions). GMP also requires that validation be associated with the rest of a process’s life cycle, however, potentially including the subsequent validation of site transfers, modified processes, and/or ongoing process verification.
It is also advisable to apply this approach to link product and process development, validate the commercial manufacturing process, and maintain the process in a controlled state as part of ongoing commercial production.
Critical process parameters (CPP), critical quality attributes (CQA), and their associated acceptance criteria should be established for validation. It is important to base these process characteristics on development data or documented process knowledge.
Process validation can be developed using a number of approaches:
- Concurrent approach
- Traditional approach
- Continuous verification approach
- Hybrid approach
Regardless of the approach employed, it is necessary to demonstrate that processes are robust and ensure consistent product quality before releasing any product to the market.
Process validation should establish that the process can consistently manage all important quality attributes and process parameters in order to ensure the validated state and acceptable product quality.
It is also necessary to clearly document the basis for identifying process parameters and quality attributes as critical or non-critical, and to consider the results of any risk assessment activities.
EU GMP Volume 4, Annex 15, states that validation should be well planned, with appropriate protocols and reporting in place, and that key elements of the site qualification and validation program are clearly defined and documented in a validation master plan (VMP) or equivalent document.4
These elements may include the specific validation approach, for example, traditional, concurrent, or continuous verification.
Qualification is a similarly documented and phased process, and it is important that qualification activities consider every stage of the process life cycle: from initial development and the user-defined specification of requirements, to the end of the use of the equipment, utilities, facilities, or systems. All acquired data should be sufficiently summarized, reviewed, and archived.
Qualification can be understood as the process of evaluating the equipment’s level of compliance with its intended use. Annex 15 requires that equipment be qualified in accordance with the environment's required characteristics, using the methodology outlined in the Annex. It is also important that the qualification (including classification) is clearly differentiated from operational monitoring.1
The concept of qualification can apply to equipment, facilities, utilities, or systems. Examples of utilities in this context are water, steam, air, or other gases, while examples of systems include premises and cleanrooms, instruments (production and QC), personnel (gowning and training), methods, and suppliers.
It is important that equipment and systems are designed, installed, qualified, maintained, operated, and monitored appropriately to ensure they function as expected and continue to support process validation.
For equipment, facilities, and utilities, it is necessary to complete and sign off on each qualification before the next one can begin. All qualifications must also be completed and implemented prior to approval of the final validation and release of the process for routine use.
Qualification documents may be combined where appropriate, for example, installation qualification (IQ) and operational qualification (OQ).4
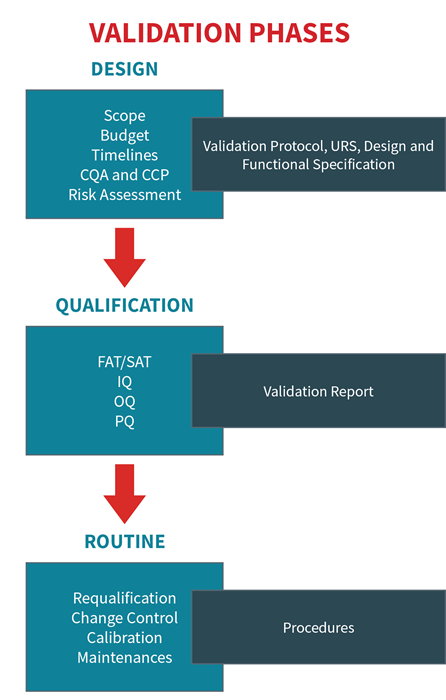
Image Credit: Particle Measuring Systems
Each qualification comprises a predetermined protocol that must be approved before work can commence. This approval is reflected in a report that records the obtained results.
Planning takes on added importance for large and complex projects, where it may be necessary to implement separate validation plans to improve clarity.4
The qualification phases are:
- User-required specification (URS)
- Design qualification (DQ)
- Factory acceptance testing (FAT)/Site acceptance testing (SAT)
- Installation qualification (IQ)
- Operational qualification (OQ)
- Performance qualification (PQ)
- Requalification
Valuable information will be collected in lab notebooks and on analytical data sheets as these concepts are applied during each validation and qualification step.
This data can be compiled and used to identify sources of variability and critical process parameters, such as temperature, pH, flow rate, and time.
The challenge in terms of validation is acquiring and collating all the process variability knowledge from the development process and applying this to statistically valid and controlled experiments that result in the delivery of well monitored, well analyzed, robust, and controlled processeSs able to produce consistent products.5
In summary, qualification is an integral part of validation. Validation and qualification must serve as the link between pharmaceutical development and routine processes, defining the means to develop a final quality product.
References and further reading
- European Commission Eudralex Volume 4: EU guidelines for good manufacturing practice for medicinal products for human and veterinary use. Annex 1: Manufacture of sterile medicinal products.
- European Medicines Agency (2017) ICH guideline Q8 (R2) on pharmaceutical development. EMA/CHMP/ICH/167068/2004. Committee for Human Medicinal Products, 22 June.
- Agalloco, J. (2004) ‘Validation: a new perspective’, in Medina, C. (ed.) Compliance handbook for pharmaceuticals, medical devices, and biologics. Boca Raton, FL: CRC Press, pp. 85–128.
- European Commission (2015) Eudralex Volume 4: EU guidelines for good manufacturing practice for medicinal products for human and veterinary use. Annex 15: Qualification and validation. Ref. Ares (2015)1380025, 30 March.
- Rathore, A.S. and Winkle, H. (2008) Quality by design, validation, and PAT: operational, statistical, and engineering perspectives. 2 August.
- United States Pharmacopeia (n.d.) <1058> Analytical instrument qualification.
Acknowledgments
Produced from materials originally authored by Serena Steidl from Particle Measuring Systems.
About Particle Measuring Systems
Particle Measuring Systems has 35 years of experience designing, manufacturing, and servicing microcontamination monitoring instrumentation and software used for detecting particles in air, liquid, and gas streams, as well as molecular contamination monitoring.
Specific applications include cleanroom monitoring, parenteral sampling, filter and in-line testing in deionized water and process chemicals, and point-of-use monitoring of inert gases and in-situ particle monitoring. Specialty monitoring includes parts cleanliness testing with a highly automated solution.
Particle measuring systems
Whether you want to protect a product or meet industry requirements, such as ISO 14644, USP 797, or GMP, Particle Measuring Systems offers a wide range of particle counters and molecular monitors to meet your needs. With 35 years of experience, it has proven reliable for application support.
Particle counters
Protect your product with PMS' reliable particle counters. It has airborne, portable, and liquid particle counters for a wide variety of applications, including DI water, chemicals, and cleanroom monitoring. Compare particle counters or learn how to monitor your cleanroom or product by reading its papers.
Molecular contamination monitors
Molecular contamination creates costly problems for high-value products, production processes, and equipment surfaces. Particle Measuring Systems has solutions for both Airborne Molecular contamination (AMC) and Surface Molecular contamination (SMC). With parts-per-trillion limits of detection, real-time sampling, NIST traceable calibrations, and various data analysis packages, you can monitor in confidence.
Gas detectors
If you need gas detectors for process control or continuous emissions monitoring, PMS can help. Get real-time, reliable results with our ammonia, hydrogen fluoride, and chlorine detectors for worker protection, CEMs, and pollution monitoring.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.net, which is to educate and inform site visitors interested in medical research, science, medical devices, and treatments.