Apoptosis is a form of programmed cell death, which occurs in cells that are irreparably damaged or in cells infected by some viruses. This mechanism is extremely important in limiting a virus’s replication and its spread to other neighboring cells.
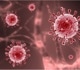
.jpg "Colorized scanning electron micrograph of an apoptotic cell (blue) infected with SARS-COV-2 virus particles (red), isolated from a patient sample. Image captured at the NIAID Integrated Research Facility (IRF) in Fort Detrick, Maryland. Image Credit: NIAID / Flickr")
Colorized scanning electron micrograph of an apoptotic cell (blue) infected with SARS-COV-2 virus particles (red), isolated from a patient sample. Image captured at the NIAID Integrated Research Facility (IRF) in Fort Detrick, Maryland. Image Credit: NIAID / Flickr
Coronaviruses
Coronaviruses are among the largest ribonucleic acid (RNA) viruses identified so far. Their genome is about 30 kb in length, with four structural proteins, and several non-structural and accessory proteins. They are classified into alpha, beta, gamma and delta coronaviruses.
Several animal and human coronaviruses have been isolated so far and mostly cause respiratory or gastrointestinal infections. Like other viruses, they hijack the host cell machinery at all stages of their replication. This involves modifying the expression of various host genes, including those that encode host defenses to facilitate viral replication.
Apoptosis
Recently, studies have shown that early in infection, coronaviruses alter cellular metabolism and increase the rate of cell death. This occurs before adaptive immunity is triggered, and this could indicate its role in productive viral infection.
Two of the most important processes involved in this modulation are apoptosis and cell cycle modification. Apoptosis is triggered by a number of signaling pathways. Apoptosis may reduce viral propagation, but the opposite effect may also occur – the release of the infectious newly produced viral particles during lytic infection.
The cell pathways that activate apoptosis can be triggered directly by the viral infection itself or indirectly via other signaling cascades. The latter is set off by the presence of the virus and recruit immune cells to the site of infection. The end event is apoptosis, causing significant damage to the tissues in lytic infection.
Apoptotic mechanism
The viral components within the cell are called pathogen-associated molecular patterns (PAMPs) and are recognized by pattern recognition receptors (PRRs). These respond by activating cytokines and other signaling molecules that upregulate interferon gene expression.
Another defense mechanism involves the recognition of viral nucleic acid by host proteins that associate, like the PRRs, with adaptor proteins to activate two key transcription factors, namely, NFĸB and IFN regulatory factor 3 (IRF3). These are translocated to the nucleus and increase the expression of type I interferons (IFNs).
These bind to IFN-α/β receptor (IFNAR), in turn, set off a system of signals involving the Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathway. The outcome is the upregulation of multiple IFN-stimulated genes (ISGs).
These genes inactivate the process involved in the synthesis of cell proteins and thus induces cell death. Another ISG that is a player in this process of shutting down translation is the combination of 2′,5′-oligoadenylate synthetase (OAS) and RNase L.
Apoptosis regulators
Apoptosis is regulated by several pro- and anti-apoptotic proteins in the host cell. One such family of proteins is the Bcl-2 (B cell lymphoma-2) family, including factors that cause poration of the mitochondrial membrane and eventual activation of caspase enzymes, initiating apoptosis. Others protect against apoptosis.
Another family is the mitogen-activated protein kinases (MAPKs), which include extracellular regulated kinase 1/2 (ERK1/2), ERK5, p38, and c-Jun N-terminal kinases (JNK). The first is upregulated by growth factors and mitogens, the last two by cell stress. The JNK pathway may lead to activation of pro-apoptotic genes or may interact with Bcl-2 genes to do so, or may activate other transcription factors like 53 and p73.
Apoptosis activation pathways
Two pathways of apoptosis have been identified, the extrinsic and the intrinsic. The former is mediated by the activation of death ligands (FasL, TNF) on their interaction with their respective death receptors (DRs). Caspase-8 is a major inducer of apoptosis in animal coronaviruses that act via DRs. This enzyme, when activated, cleaves Bid, a Bcl2 protein, leading to the release of mitochondrial cytochrome c. The latter is key to the formation of the apoptosome with the cleavage and activation of caspase 9.
The intrinsic pathway is based on the permeabilization of mitochondrial outer membranes and is chiefly seen with animal coronaviruses. Bcl2 proteins like Bax and Bak favor apoptosis and are required to trigger this process via the intrinsic or mitochondrial-mediated pathway, along with caspase-9.
Many animal coronaviruses cause activation of both extrinsic and intrinsic pathways, showing the intricate network of signaling pathways involved in apoptosis.
Coronavirus interactions with apoptosis genes
Viral genomes often encode genes that are homologous to host anti-apoptotic proteins. This allows them to delay apoptosis in the early stages of infection and thus generate an adequate yield of new virions. Other viral proteins can prevent the transcription of some apoptotic genes or cause the modification of their protein products, to suppress their activity.
The induction of apoptosis by viral proteins may involve the termination of cap-dependent translation in host cells, inducing apoptosis, as seen with the poliovirus. In the case of the hepatitis C virus, the viral NS3 protein interacts with caspase-8, to trigger cell death.
The current review is focused on identifying pro-apoptotic mechanisms triggered by animal coronaviruses. For instance, the Porcine Epidemic Diarrhea Virus (PEDV) triggers changes in the expression of 14 genes involved in caspase-independent apoptosis to promote its replication, as well as producing the characteristic features of disease.
Another way in which apoptosis is involved in viral replication is by activating caspase, which is required for the cleavage of the viral nucleocapsid (N) protein during this process. This is the case with the transmissible gastroenteritis virus (TGEV), which activates the MAPK family, as well as FasL- and mitochondria-induced apoptosis. This process also includes the downregulation of host microRNAs that suppress the mitochondrial apoptotic pathway.
Betacoronaviruses, such as the murine hepatitis virus (MHV), induces apoptosis. This virus shows envelope protein (E)-induced apoptosis, again via caspase activation. In this case, apoptosis appears to be protective against virion production.
With the porcine hemagglutinating encephalomyelitis virus (PHEV), cell death is mediated by DRs and the mitochondrial-mediated pathways, involving caspase-8 and caspase-9, as initiators, and caspase-3, as an effector of apoptosis.
Similarly, the gamma- and delta-coronaviruses show complex induction of cellular signaling pathways in the host cell, leading to the modulation of apoptosis. Thus, many viral proteins modulate host cell pathways to promote viral replication and disease features.
What are the implications?
The fact that apoptosis occurs following infection of multiple hosts by a range of coronaviruses leads to the conclusion that it is a common feature of infection by human and animal coronaviruses and aims to favor propagation.
With the earlier severe acute respiratory syndrome coronavirus (SARS-CoV), the 7a protein forms a complex with Bcl-XL protein or other pro-survival factors, inactivating them and thus allowing apoptosis to proceed via caspase-3 activation. The 3CL-like protease (3CLpro) also activates both caspase-3 and caspase-9 to induce apoptosis in cell culture.
Both SARS-CoV and the currently circulating SARS-CoV-2 viruses have the ORF3a protein that triggers cell death via caspase activation in the extrinsic pathway through DRs.
A similar role has not yet been shown for viral proteins in animal coronavirus infections. However, with PEDV, the S1 subunit of the spike protein, and the non-structural protein 1 (NSP1), has been suggested to be the initiator and effector of cell death, respectively, based on recent experiments.
More studies may help understand how these pathways operate in other coronaviruses. The current state of knowledge may indicate the utility of targeting this pathway for managing coronavirus infections.
 Scientists find that human embryos are vulnerable to COVID
Scientists find that human embryos are vulnerable to COVID