Single guide ribonucleic acid (sgRNA) molecules are produced by discontinuous transcription, in which viral RNA-dependant RNA polymerase pauses early negative-sense RNA synthesis and then jumps to the other end of the genome. The specifics of this process are still not fully understood.
Since sgRNAs can play an important role in the pathogenicity of viruses, including the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), many researchers are currently exploring this process. To this end, a recent study published on the preprint server bioRxiv* discusses a bioinformatics method to help identify and explore sgRNAs uncovered in next-generation sequencing (NGS) data.
 Study: sgDI-tector: defective interfering viral genome bioinformatics for detection of coronavirus subgenomic RNAs. Image Credit: Connect world / Shutterstock.com
Study: sgDI-tector: defective interfering viral genome bioinformatics for detection of coronavirus subgenomic RNAs. Image Credit: Connect world / Shutterstock.com
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
About the study
In the current study, the researchers generated NGS data on RNA isolated from human HEK cells transduced with angiotensin-converting enzyme (ACE) and subsequently infected with SARS-CoV-2. DI-tector, which is the user-friendly bioinformatic pipeline, was then used to run the RNAseq data to characterize and quantify defective viral genomes (DVG)s.
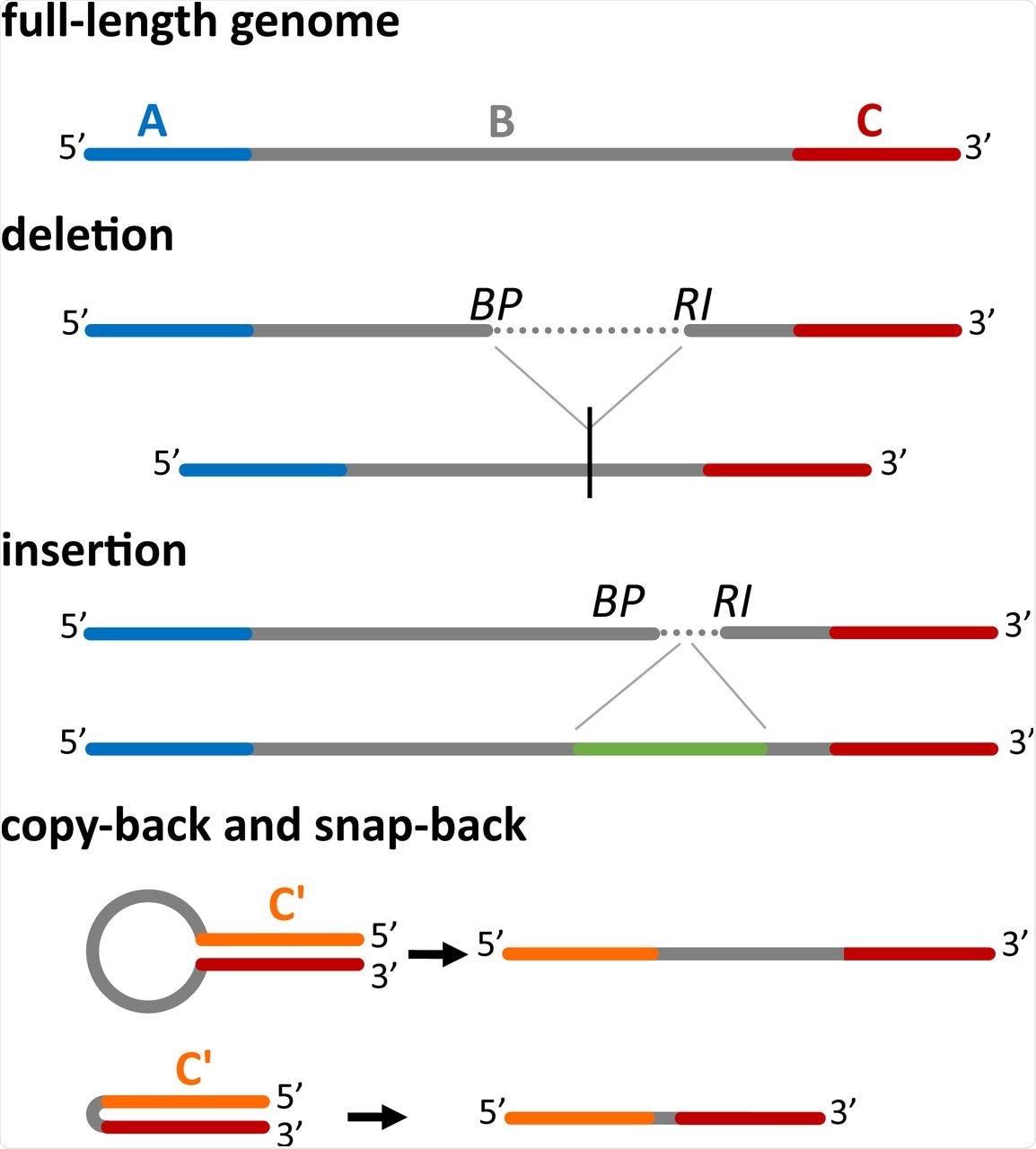 Four main classes of DI genomes can be detected by DI-tector. The full-length genome (divided here in three regions, A, B and C) is shown first. When a part of the region B is missed then DI-tector detects a deletion, if instead a region is added, DI-tector detects an insertion. Copy-backs and snap-backs are formed through junctions involving the two strands (positive-sense and negative-sense). C’ is the region complementary to C. BP: breakpoint site, RI: reinitiation site.
Four main classes of DI genomes can be detected by DI-tector. The full-length genome (divided here in three regions, A, B and C) is shown first. When a part of the region B is missed then DI-tector detects a deletion, if instead a region is added, DI-tector detects an insertion. Copy-backs and snap-backs are formed through junctions involving the two strands (positive-sense and negative-sense). C’ is the region complementary to C. BP: breakpoint site, RI: reinitiation site.
The researchers observed between 33-40% of reads mapped to the SARS-CoV-2 genome and a much smaller quantity of DVG reads, the most common of which were deletions. This suggests that DI-tector results can be used to quantify viral sgRNA data from NGS. The second most common DVG reads were insertions, followed by copy-back or snap-back results.
Leader-body junctions are detected by the DI-tector as deletion DVGs. These are formed during the transcription of sgRNAs.
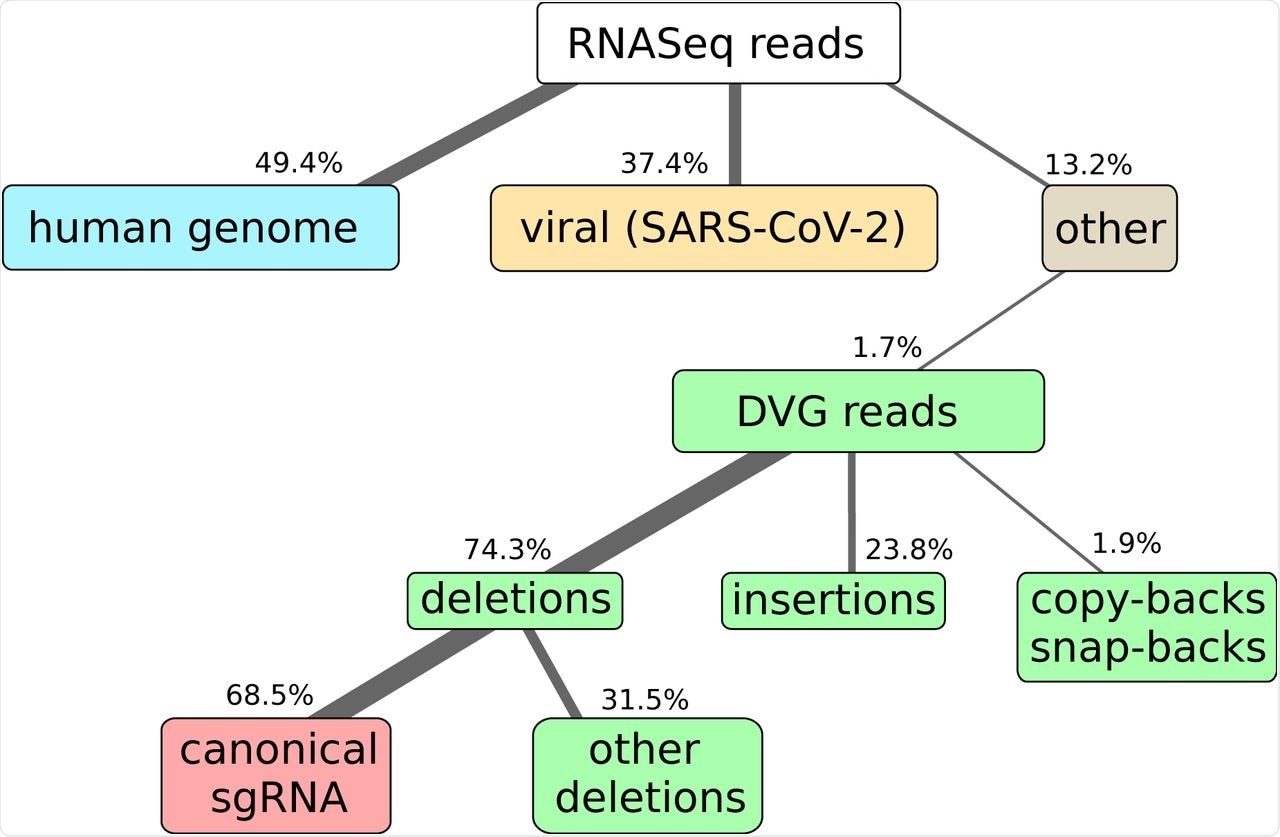 Here we show the results of RNAseq and alignment of the reads to the human and SARS-CoV-2 genomes. NGS library preparation was performed with an ribodepletion step. The unmapped reads have been further processed with DI-tector and the resulting characterization of the DVG reads into deletions, insertions and copy-backs/snap-backs is given. The percentage of junction reads corresponding to canonical sgRNA, that is standard annotated subgenomic ORFs for SARS-CoV-2 (S, 3A, E, M, 6, 7a, 7b, N, 10), is specified. All percentages are averaged over three biological replicates. Cells colors are given to classify reads in: host-related reads (blue), viral reads (yellow), DVG reads (green), canonical sgRNA reads (red), other reads (gray).
Here we show the results of RNAseq and alignment of the reads to the human and SARS-CoV-2 genomes. NGS library preparation was performed with an ribodepletion step. The unmapped reads have been further processed with DI-tector and the resulting characterization of the DVG reads into deletions, insertions and copy-backs/snap-backs is given. The percentage of junction reads corresponding to canonical sgRNA, that is standard annotated subgenomic ORFs for SARS-CoV-2 (S, 3A, E, M, 6, 7a, 7b, N, 10), is specified. All percentages are averaged over three biological replicates. Cells colors are given to classify reads in: host-related reads (blue), viral reads (yellow), DVG reads (green), canonical sgRNA reads (red), other reads (gray).
The pipeline that the scientists have produced starts from the DI-tector output and is based on the assumption that sgRNA coding for expressed open reading frames (ORFs) are more frequently transcribed and that there is a leader sequence shared between sgRNAs. The leader/junction TRS is not needed to apply the algorithm.
When the researchers applied their algorithm, they observed clear signals of several ORFs, as well as signals of direct transcription of ORF 7b. As well as this, the researchers observed a leader-body junction that could result in the expression of a non-canonical ORF.
The number of counts for this was comparable with ORF E and larger than ORF 7b. The non-canonical ORF was named U6bc1.
Additional leader-body junctions gave rise to more ORFs. In order to control for false positive, the RNASeq data from mock-infected HEK-ACE2 cells were analyzed. The sgDI-tector did not give any sgRNA read, further confirming the new approach.
Reverse-transcriptase qualitative polymerase chain reaction (RT-qPCR) was used to further test the accuracy of the algorithm. The non-canonical ORF U3dc4 was validated. This ORF was less present than the other non-canonical ORFs and has been previously detected by STAR approach, but not by any conventional methods.
RT-qPCR analysis revealed this ORF was only detected in RNA extracted from cells similar to the detection of canonical ORF encoding the nucleocapsid (N) protein, thereby validating the presence of the ORF and confirming that the sgDI-tector can help scientists understand sgRNA populations.
Pre-existing techniques to detect sgRNA expression levels in coronaviruses include reads per kilobase of transcript per million mapped reads (RPKM). This approach does not produce results that correlate with the junction analysis performed by the sgDI-tector. Furthermore, this decumulation results in several ORFs having a negative number of events.
The authors argue that the lack of correlation is due to the failure of the RPKM method, as the same pipeline shows a much higher correlation between junction counts and decumulation of RPKM in a separate dataset.
The sgDI-tector does not require the operator to know the identity of the leader sequence, as the junction-spanning reads are used to recover its position. Focusing on the nucleotides around the RI positions for the reads detected by the DItector allows the tools to discover the leader-body conserved transcription regulatory sequence (TRS).
A previously reported TRS 5’-ACGAAC-3’ is present with most cases and in the junctions with the highest amounts of observed counts. The nucleotides in positions 71-72-73 are perfectly conserved within the body partners of the analyzed leader-body junctions.
Conclusions
The authors have shown that their method results in a strong correlation between NGS data analyzed by Spliced Transcripts Alignment to a Reference (STAR) algorithms, as well as data analyzed by their sgDI-tector method. This method does not need the leader sequence input and has been specifically designed for addressing sgRNA level expression, thus allowing it to work without the need for unconventional parameter choice.
The approach discussed in the current study is user-friendly and does not require the same fine-tuning STAR algorithms do in order to analyze sgRNA data. This new method could prove invaluable to researchers investigating sgRNA, which could be particularly useful in the development of new techniques and medications against SARS-CoV-2.
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
Di Gioacchino, A., Legendere, R., Rahou, Y., et al. (2021). sgDI-tector: defective interfering viral genome bioinformatics for detection of coronavirus subgenomic RNAs. bioRxiv. doi:10.1101/2021.11.30.470527. https://www.biorxiv.org/content/10.1101/2021.11.30.470527v1
- Peer reviewed and published scientific report.
Di Gioacchino, Andrea, Rachel Legendre, Yannis Rahou, Valérie Najburg, Pierre Charneau, Benjamin D. Greenbaum, Frédéric Tangy, Sylvie van der Werf, Simona Cocco, and Anastassia V. Komarova. 2021. “SgDI-Tector: Defective Interfering Viral Genome Bioinformatics for Detection of Coronavirus Subgenomic RNAs.” RNA 28 (3): 277–89. https://doi.org/10.1261/rna.078969.121. https://rnajournal.cshlp.org/content/28/3/277.
 Scientists find that human embryos are vulnerable to COVID
Scientists find that human embryos are vulnerable to COVID