The discovery of new and more transmissible variants of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has been ongoing since the start of the coronavirus disease 2019 (COVID-19). Therefore, it is crucial to promptly identify mutations in the virus as that could aid immensely in outbreak control efforts as well as shed light on the new variants that should be monitored more closely.
 Study: Inferring effects of mutations on SARS-CoV-2 transmission from genomic surveillance data. Image Credit: NIAID
Study: Inferring effects of mutations on SARS-CoV-2 transmission from genomic surveillance data. Image Credit: NIAID
 *Important notice: medRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
*Important notice: medRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
Background
Understanding mutations in viruses is vital as that could help us understand how efficiently they infect hosts and inform public health policies to control the spread of the virus. However, estimating how individual mutations affect viral transmission is not easy.
Currently, phylogenetic analyses or fitting changes in variant frequencies to a simple growth model are the techniques used to estimate changes in viral transmission. The former can be challenging owing to a high degree of sequence similarity. Another drawback of phylogenetic analyses is that they rely heavily on Markov chain Monte Carlo sampling, making it difficult to track large datasets. In terms of simple growth models, they lack the ability to account for competition between multiple variants. The above two methods also do not account for the superspreading or travel of infected individuals.
A New Study
To overcome the above drawbacks, scientists developed a novel SIR-based method to gain insights into the effects of single nucleotide variants (SNVs) on viral transmission from genomic surveillance data. The study also accounted for factors, such as competition between viral lineages, travel, superspreading, etc.
Simulations showed that this approach could reliably estimate transmission effects of SNVs even from limited data – a huge advantage. The method was applied to more than 1.6 million SARS-CoV-2 sequences from 87 geographical regions. The goal was to understand the effects of mutations on viral transmission throughout the pandemic.
Researchers also quantified the influence of travel and competition between multiple variants and found that travel only slightly affected the estimated changes in transmission. However, significant effects of competition between variants were observed.
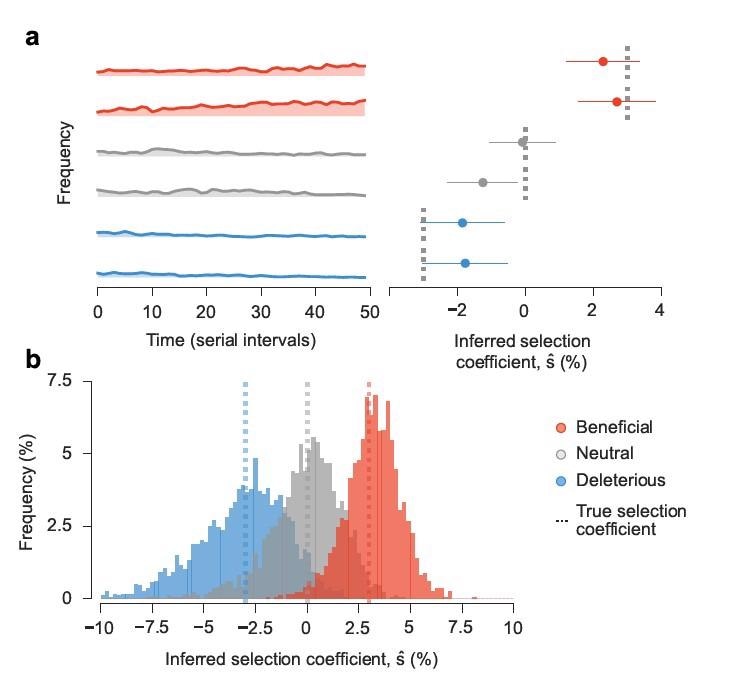
Approach accurately estimates transmission effects of mutations in simulations. Simulated epidemiological dynamics beginning with a mixed population containing variants with beneficial, neutral, and deleterious mutations. a, Selection coefficients for individual SNVs, shown as mean values ± one theoretical s.d., can be accurately inferred from stochastic dynamics in a typical simulation (Methods). b, Extensive tests on 1,000 replicate simulations with identical parameters show that inferred selection coefficients are centered around their true values. Deleterious coefficients are slightly more challenging to accurately infer due to their low frequencies in data. Simulation parameters. The initial population is a mixture of two variants with beneficial SNVs (s = 0.03), two with neutral SNVs (s=0), and two with deleterious SNVs (s=−0.03). The number of newly infected individuals per serial interval rises rapidly from 6,000 to around 10,000 and stays nearly constant thereafter. Dispersion parameter k is fixed at 0.1.
Main Findings
Scientists applied SARS-CoV2 data across many regions and revealed multiple mutations that strongly affect the transmission rate. These mutations were found both within and outside the Spike protein. In the current study, researchers also focussed on travel and competition across variants (using the history of 20E (EU1) as an example) – factors not well accounted for in previously used methods. They quantified the impacts of travel and competition between different lineages on the inferred transmission effects of mutations.
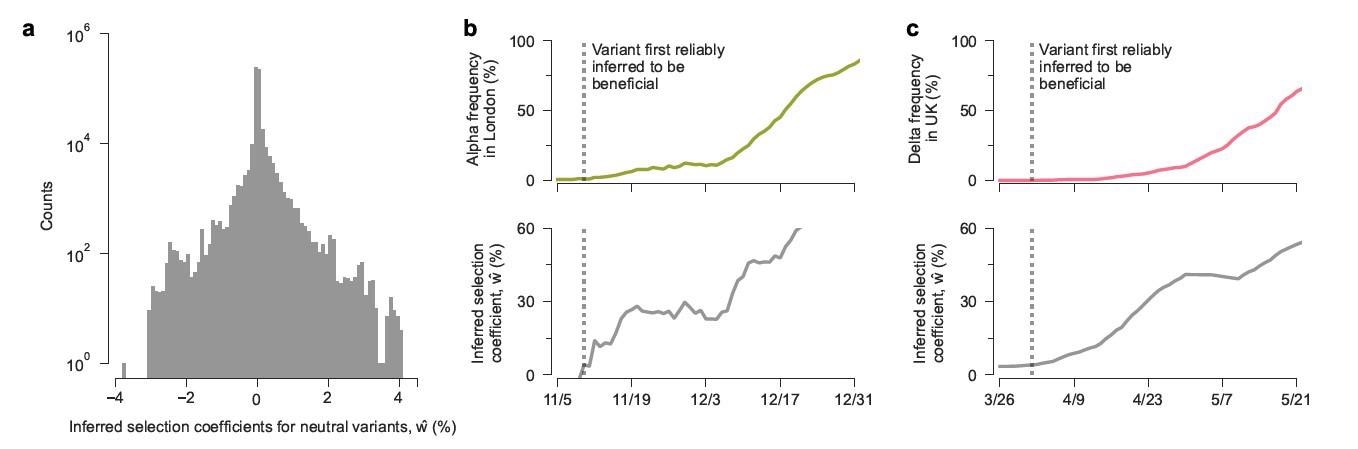
An encouraging observation was that the model was capable of detecting lineages with increased transmission as they arise. Significant transmission advantages were inferred within a week of their appearances in regional data regarding the Alpha and Delta variants. At this time, the regional frequencies of the Alpha and Delta variants were 1%. While the data in the study only extended to August 6th, 2021, researchers would estimate a selection coefficient of 55.2% for the newly emerged Omicron variant, based on the mutations that it shares with the previous variants. Therefore, the model enables the quick identification of variants and mutations that could affect transmission from genomic surveillance data, thereby providing an “early morning” for more transmissible variants.
Concluding Remarks
Scientists have stated that sustained research is essential to identify and characterize new variants as they emerge. An example is the newly emerged Omicron variant in South Africa. The model developed in this study has focused exclusively on SARS-CoV-2; however, it could be applied to study the transmission of other pathogens such as influenza. The model, together with extensive genomic surveillance data, is a powerful method for promptly identifying more transmissible viral variants and, subsequently, quantifying the contributions of individual mutations to changes in the transmission rates.
*Important notice: medRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
 Scientists develop a dual action strategy to destabilize the COVID virus
Scientists develop a dual action strategy to destabilize the COVID virus