Human monkeypox is a zoonotic disease that originated in the central and west African jungles and was first reported in the Democratic Republic of Congo in 1970. Monkeys and rodents spread the disease, although the original reservoir remains unknown. Humans contract MPXV infection through contact with infected animals/humans and contaminated materials. MPXV is an enveloped virus with approximately 197 kb of double-stranded (ds) DNA.
MPXV belongs to the Orthopoxvirus genus under the Poxviridae family of double-stranded DNA viruses. Genome sequencing has identified two clades of MPXV: Congo Basin (CB) and West African (WA) clades. The CB clade causes severe disease and higher mortality. Generally, poxviruses (including MPXV) exhibit greater tolerance to a wide range of pH and are much more resistant to desiccation than other enveloped viruses. Besides, they are less sensitive to disinfectants than other enveloped viruses.
 Study: A new and efficient enrichment method for metagenomic sequencing of monkeypox virus Image Credit: NIAID
Study: A new and efficient enrichment method for metagenomic sequencing of monkeypox virus Image Credit: NIAID
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
About the study
In the present study, researchers evaluated a new method of MPXV DNA enrichment in comparison to the non-enrichment approach. Clinical specimens from lesions or vesicular fluid swabs were obtained, and two MPXV-positive samples (MP01 and MP03) were selected. DNA extraction was performed by two different protocols.
In the first (non-enrichment) method, DNA was extracted using MagNA pure compact nucleic acid isolation kit I. The second protocol was modified from a saponin-based differential lysis technique and centrifuged at high g-force (35,000 g). This method was designed to enrich MPXV DNA samples to avoid wasting sequencing quota.
A soft centrifugation step (at low g-force) was performed initially to remove large particles. Host DNA was depleted with the treatment of saponin, sodium chloride (NaCl), and deoxyribonuclease (DNase). NaCl, DNase, and saponin were removed for sequencing. The samples were processed using the non-enrichment (MP01bCHUAC, MP03bCHUAC) and enrichment methods (MP01CHUAC, MP03CHUAC).
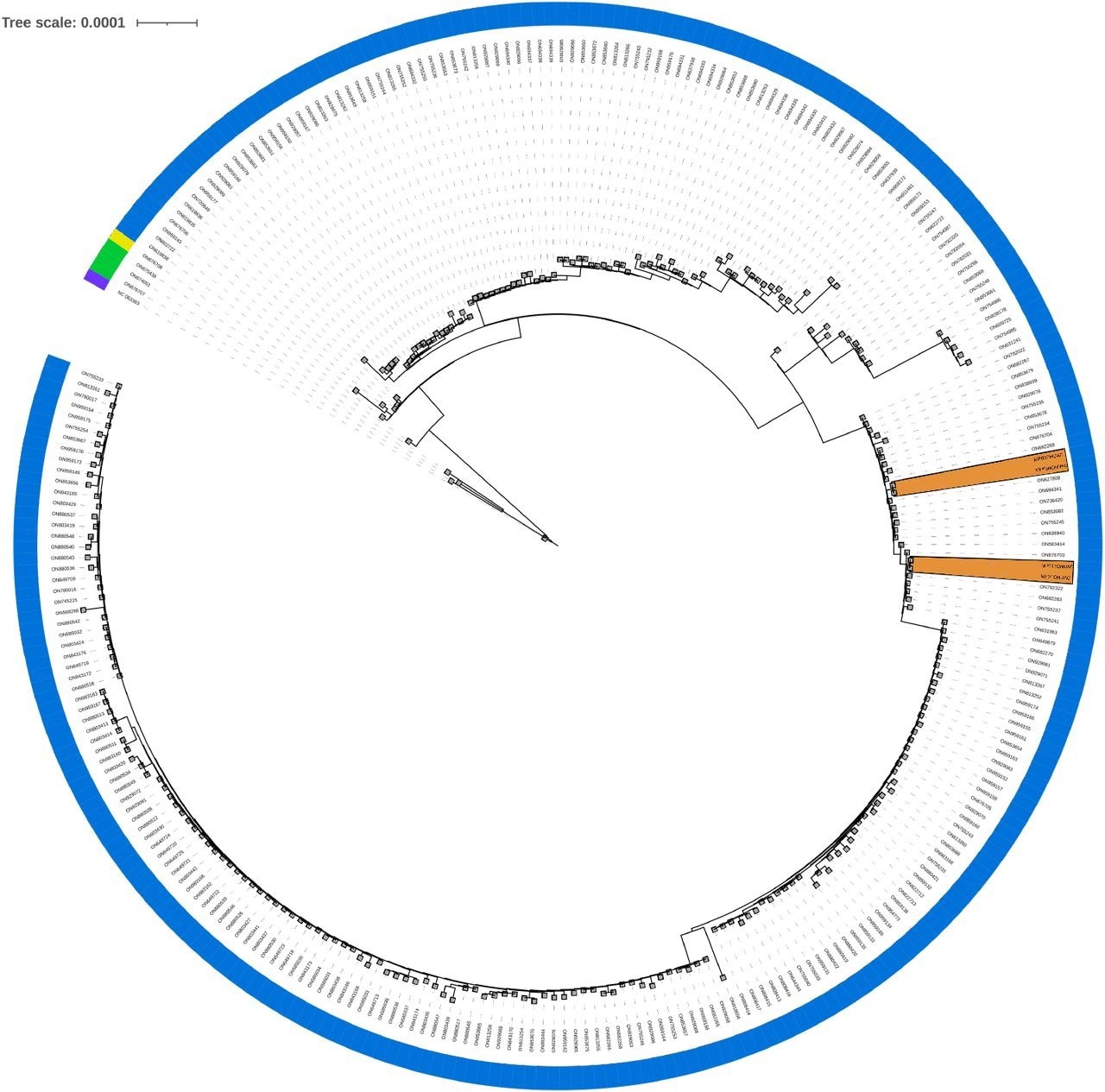 Phylogenomic tree. Phylogenomic analysis of the present study’s samples, comparing them to all complete MPXV genomes available in GenBank to date (2022-07-18, 275 genomes from taxid 10244). This does not pretend to infer the evolution of the virus, only to locate the most similar entries to the samples in this study. A color strip indicates each sample’s lineage (A: purple, A.1.1: yellow, A.2: green, B.1: blue), and orange areas highlight the study’s samples.
Phylogenomic tree. Phylogenomic analysis of the present study’s samples, comparing them to all complete MPXV genomes available in GenBank to date (2022-07-18, 275 genomes from taxid 10244). This does not pretend to infer the evolution of the virus, only to locate the most similar entries to the samples in this study. A color strip indicates each sample’s lineage (A: purple, A.1.1: yellow, A.2: green, B.1: blue), and orange areas highlight the study’s samples.
Findings
In the preliminary results via the Kraken 2 taxonomic classification system using raw unfiltered reads, the authors noted that most reads in the enriched samples (MP01CHUAC and MP03CHUAC) belonged to MPXV, and almost none were classified as host contamination. In contrast, most reads in non-enriched samples (MP01bCHUAC and MP03bCHUAC) were of human DNA.
When Best Match Tagger (BMTagger) removed human DNA contamination, the read counts for MP01CHUAC and MP03CHUAC reduced by 1% to 2 %, while the reduction for MP01bCHUAC and MP03bCHUAC was 90% to 95%. In the subsequent step of quality control, any of the reads not classified as Orthopoxvirus was removed. This resulted in less drastic change and read counts decreased by 30% for enriched samples and 8% - 41% for non-enriched samples.
Compared to raw/original reads, read counts after the final quality control step decreased by 50% for enriched samples and 93% - 98% for non-enriched samples. The remaining reads were aligned to a reference sequence where reads from enriched samples had a median depth of 1500 – 1800, while those from non-enriched samples had a median depth of 80 – 100.
Nevertheless, all samples produced a quality consensus sequence of the MPXV B.1 lineage. MP01CHUAC had one nucleotide mutation, whereas MP03CHUAC had two amino acid substitutions against the reference genome.
Conclusions
The study found significant differences between both protocols when comparing depth and read counts. The decrease in read counts when host reads were removed was massive (90% to 95%) for non-enrichment protocol but marginal (1% to 2%) for enrichment protocol. In addition, the enrichment method retained half the reads after the final quality control step, while the non-enrichment protocol kept only 2% - 3% of the original reads.
After aligning the cleaned reads to a reference MPXV sequence, the median depth was higher for enriched samples than for non-enriched samples. All samples generated good quality consensus based on alignment, but the increased depth (with enrichment protocol) is vital to trust the observed genetic changes.
In conclusion, the new enrichment method significantly improved sequencing efficiency, number of viral reads, depth, and the trustworthiness of consensus sequences. Notably, removing host sequences before sequencing allows for the inclusion of more samples per cartridge, reducing costs and time for diagnosis and increasing the efficiency of sequencing.
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
Aja-Macaya P, Rumbo-Feal S, Poza M, Canizares A, Vallejo JA, Bou G. A new and efficient enrichment method for metagenomic sequencing of monkeypox virus. medRxiv, 2022, DOI: 10.1101/2022.07.29.22278145, https://www.medrxiv.org/content/10.1101/2022.07.29.22278145v1
- Peer reviewed and published scientific report.
Aja-Macaya, Pablo, Soraya Rumbo-Feal, Margarita Poza, Angelina Cañizares, Juan A. Vallejo, and Germán Bou. 2023. “A New and Efficient Enrichment Method for Metagenomic Sequencing of Monkeypox Virus.” BMC Genomics 24 (1). https://doi.org/10.1186/s12864-023-09114-w. https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-023-09114-w.
 Infectious H5N1 virus detected in dairy farm air
Infectious H5N1 virus detected in dairy farm air