Several fat sum parameters or fat indices are utilized for the characterization and quality control of fats and oils since establishing the exact content of individual glycerides in fats and oils is time-consuming and challenging. Oils and fats are not only important for cooking, they are also a crucial ingredient in personal care products and pharmaceuticals, like creams and ointments.
Hence, several standards and norms describe the calculation of the most crucial quality control parameters. This article outlines eight vital analytical techniques for the below fat parameters in edible fats and oils:
- Hydroxyl value according to
- Iodine value based on
- EN ISO 3961,
- ASTM D5554,
- AOAC 920.159,
- AOAC 993.20,
- USP<401> Method II,
- Ph.Eur. 2.5.4 Method B,
- AOCS Cd 1d-92
- Peroxide value according to
- EN ISO 27107,
- EN ISO 3960,
- AOAC 965.33,
- Ph.Eur. 2.5.5,
- USP<401>
- Water content according to
- EN ISO 8534,
- GB/T 26626,
- AOAC 984.20
- Saponification value according to:
- EN ISO 3657,
- ASTM D5558,
- AOAC 920.160,
- USP<401>,
- Ph.Eur. 2.5.6
- Oxidation stability according to
- AOCS Cd 12b-92,
- EN ISO 6886,
- GB/T 21121,
- JOCS 2.5.1.2
- Acid value, free fatty acids (FFA) according to
- EN ISO 660,
- USP<401> Method I,
- Ph.Eur. 2.5.1
- Nickel traces using polarography
Extra care is taken to avoid chlorinated solvents in these techniques. Additionally, as many of the mentioned techniques as possible are automated.
Water Content
Summary
Coulometric Karl Fischer is a favored technique for this analysis due to the low water content of pure fats and oils. Volumetric Karl Fischer titration is recommended for margarine and butters which possess relatively high water contents.
Instruments
- Coulometric KF titrator or
- Volumetric KF titrator
Electrodes
| . |
. |
| Double Pt-wire electrode for coulometry |
6.0341.100 |
| Generator electrode with diaphragm |
6.0344.100 |
Volumetric
| . |
. |
| Double Pt-wire electrode for volumetry |
6.0338.100 |
Reagents
Coulometric
- Hydranal Coulomat Oil or equivalent
- Hydranal Coulomat CG or equivalent
Volumetric
- Hydranal Composite 2 or Hydranal Composite 1 or equivalent
- Methanol, dry, p.a.
- 1-Decanol, p.a.
Solutions
| . |
. |
| Solvent mixture |
Methanol / 1-decanol, Φ(MeOH) = 66% (v/v) |
Standard
- Standard with a water content of 10 mg/g
Sample Preparation
Before adding them into the titration vessel, hard fats should be melted. As the water is inhomogeneously distributed in them, margarine and butter should be homogenized first. They should not be heated above 25 °C, or phase separation may occur.
Analysis
Sample (Coulometric)
Around 100 mL of coulometric reagent is added into the titration cell and conditioned until a constant drift is acquired (< 10 μg/min is usual). A syringe is rinsed three times with the sample (approx. 1 mL) and then this sample is discarded. The syringe is filled with sample, and an appropriate volume of sample (as shown in the table below) is added to the titration cell and titrated to the end point.
Table 1: Sample sizes dependent on the expected water content according to EN ISO 8534
| Expected water content / % |
Minimal sample amount / g |
| 0.0001 |
10 |
| 0.001 |
10 |
| 0.01 |
5 |
| 0.1 |
2 |
| 1 |
0.2 |
Titer (Volumetric)
Around 30 mL of solvent mixture is added into the titration cell and conditioned until a constant drift of approximately 10–20 μL/min is attained. The water standard is put into a dry syringe and around 1 g of water standard is added into the titration cell and titrated immediately to the end point.
Sample (Volumetric)
Around 30 mL of solvent mixture is added into the titration cell and then conditioned until a constant drift of approximately 10–20 μL/min is acquired. The sample is then filled into a dry syringe (without needle). A suitable volume of sample (see below) is added to the titration cell and then titrated to the end point.
Table 2: Sample sizes dependent on the expected water content according to EN ISO 8534.
| Expected water content / % |
Minimal sample amount / g |
| 0.01* |
20 |
| 0.1* |
5 |
| 1 |
1 |
| 5 |
0.2 |
| 10 |
0.1 |
| 20 |
0.05 |
* For these water contents Metrohm recommends the coulometric Karl Fischer titration.
Parameters
Coulometric
| . |
. |
| Mode |
KFC |
| Start drift |
20 μg/min |
| EP at |
50 mV |
| Dynamics |
70 mV |
| Min. rate |
15 μg/min |
| Stop criterion |
Rel. drift |
| Rel. stop drift |
5 μg/min |
| Extraction time |
0 s |
Volumetric (titer and sample)
| . |
. |
| Mode |
KFT Ipol |
| Start drift |
20 μL/min |
| EP at |
250.0 mV |
| Dynamics |
100 mV |
| Stop criterion |
Drift |
| Stop drift |
20 μL/min |
| Extraction time |
0 s |
Calculations

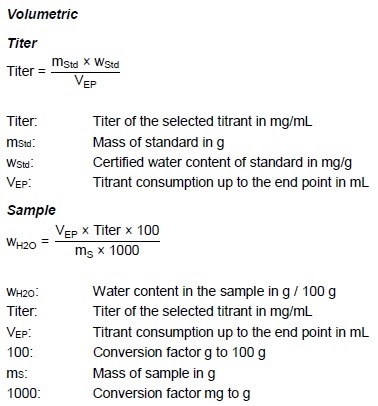
Comments
- EN ISO 8534 recommends volumetric Karl Fischer titration for samples with a water content of 1 to 100 mg of water. Coulometric Karl Fischer titration is recommended for samples with a water content of 10 µg to 10 mg of water.
- For fats and oils with a water content of 1% or lower, coulometric Karl Fischer titration is recommended as a greater precision can be achieved.
- The method differs from EN ISO 8534 in the following way:
- A mixture of methanol and 1-decanol is used as solvent instead of a mixture of methanol and chloroform.
- For the preparation of the syringe for the titer determination, see Metrohm Application Bulletin AB424
- The added sample weight is determined by weighing the full syringe before the addition of the sample and then again after the addition of the sample into the titration cell. The difference is determined and used as a sample weight.
References
- Application Bulletin AB-424 Titer determination in volumetric Karl Fischer titration
- EN ISO 8534 Animal and vegetable fats and oils – determination of water content – Karl Fischer method (pyridine free)
- GB/T 26626 Animal and vegetable fats and oils – Determination of water content – Karl Fischer method
- AOAC 984.20 Moisture in oils and fats. Karl Fischer method
Oxidation Stability
Summary
The Rancimat technique is an accelerated aging test. Air passes through the sample in the reaction vessel at a constant elevated temperature, and fatty acids are oxidized in this process. When the test is finished, volatile secondary reaction products are formed, which are carried into the measuring vessel via the air stream and absorbed in the measuring solution (deionized water).
This leads to a spike in the electrical conductivity, which is recorded continuously. The time until secondary reaction products are formed and detected is known as induction time, this characterizes the oxidation stability of fats and oils.
Instruments
- Rancimat
- Equipment for determining the temperature correction
Reagents
Sample Preparation
For this technique no sample preparation is needed. Liquid oils can be weighed indirectly. The sample can be previously melted in a water bath in case of problems such as weighing solid fat into the bottom part of the reaction vessel. Care must be taken that the temperature of the water bath is not significantly higher than the melting point of the sample. If not, deterioration of the sample can be anticipated.
Analysis
The temperature of the heating block must be stable before the determination can start. Fill each measuring vessel with 60 mL deionized water and place it on the Rancimat with the measuring vessel cover and the integrated conductivity cell.
Next, utilize a clean, new reaction vessel and weigh 3 g of sample into the bottom part and close it with the reaction vessel cover with the air inlet tube attached. Connect the two tubings for the air supply, place the reaction vessel in the heating block, and begin data recording immediately.
Parameters
| . |
. |
| Sample size |
3 g |
| Measuring solution |
60 mL deionized water |
| Temperature |
80 – 160 °C |
| Gas flow |
20 L/h |
| Evaluation |
Induction time |
Example Determination
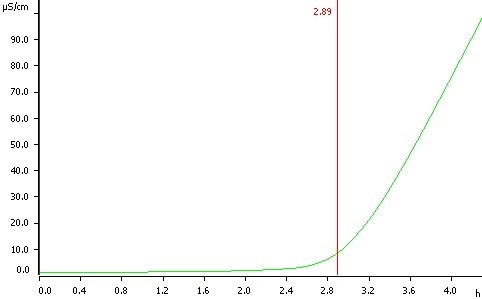
Figure 1: Determination of oxidation stability of sunflower oil at a temperature of 120 °C, induction time 2.89 h.
Typical Results
| Sample |
Temperature/°C |
Induction time/h |
| Corn oil |
120 |
approx. 5 |
| Hazelnut fat |
120 |
10 – 12 |
| Hazelnut oil |
120 |
7 – 11 |
| Lard |
100 |
1 – 3 |
| Linseed oil |
110 |
0.5 – 2 |
| Margarine |
120 |
2 – .6 |
| Olive oil |
120 |
6 – 11 |
| Palm oil |
120 |
7 – 12 |
| Peanut fat |
120 |
9 – 10 |
| Peanut oil |
120 |
3 – 15 |
| Pumpkin seed oil |
120 |
approx. 7 |
| Rapeseed oil |
120 |
3 – 5 |
| Safflower oil |
120 |
1 – 2 |
| Sesame oil |
120 |
approx. 5 |
| Soybean oil |
120 |
1 – 7 |
| Sunflower oil |
120 |
1 – 4 |
| Tallow |
120 |
3 – 8 |
Comments
- For each determination, it is advisable to utilize a new reaction vessel to avoid side reactions because of contamination. The reaction vessel is air-cleaned inside and out by a sharp stream of nitrogen before the sample is weighed in to remove particles (e.g. from the cardboard box).
- The most important parameter in this application is temperature. So, a temperature correction must be incorporated into the technique settings to compensate for the cooling because of the gas flow. In the manual of the instrument software tabled values are available for different gas flow rates and temperatures.
It is recommended to determine the temperature correction by utilizing the optional equipment for determining the temperature correction for the best reproducibility of results. See the instructions for the use of the instrument for further information.
- The induction time is typically determined at 120 °C. The temperature can be set in a way that the induction time is within 4 to 10 hours. When the temperature is increased by 10 °C, the induction time generally decreases by a factor of 2 and vice versa.
References
- AOCS Cd 12b-92 Sampling and analysis of commercial fats and oils – Oil Stability Index (OSI)
- EN ISO 6886 Animal and vegetable fats and oils – determination of oxidative stability (accelerated oxidation test)
- GB/T 21121 Animal and vegetable fats and oils – Determination of oxidation stability
- JOCS 2.5.1.2 Oxidation stability - CDM (Conductometric Determination Method)
- Metrohm Application Bulletin 204 Oxidation stability of oils and fats – Rancimat method
Iodine Value
Summary
For the characterization of oils and fats, and as a quality control parameter, the iodine number is utilized. As a sum parameter, it permits quantification of the amount of unsaturated fatty acids present in oils and fats. The more unsaturated fatty acids, the more iodine reacts with the double bonds, resulting in a higher iodine value.
Typically, after the addition of the reaction solution (Wijs solution), the samples must be placed in the dark for up to two hours. Below, we outline an altered analysis based on AOAC 920.159, AOAC 993.20, EN ISO 3961, ASTM D5554, and AOCS Cd 1d-92. Magnesium acetate is added as a catalyst for this modified analysis, decreasing the reaction time from up to 2 hours to 5 minutes.
Instruments
- Sample changer with Swing Head and DIS-Cover
- Titrator with DET mode
- 2x Burette 20 mL (Glacial acetic acid, Mg(CH3COO)2)
- 4x Burette 50 mL (H2SO4, ICl, KI, Na2S2O3)
- Propeller Stirrer
Electrodes
| . |
. |
| iPt Titrode |
6.0471.300 |
Reagents
- Sulfuric acid, c(H2SO4) = 0.5 mol/L, volumetric solution
- Potassium iodate, KIO3, p.a.
- Potassium iodide, KI, p.a.
- Sodium thiosulfate, c(Na2S2O3) = 0.1 mol/L, volumetric solution
- Magnesium acetate, Mg(CH3COO)2, purum
- Glacial acetic acid, p.a.
- Iodine chloride, Wijs-solution, c(ICl) = 0.1 mol/L, volumetric solution
Solutions
| . |
. |
| Titrant |
c(Na2S2O3) = 0.1 mol/L
If possible this solution should be bought from a supplier. |
| Potassium iodide solution |
β(KI) = 100 g/L
50 g potassium iodide is weighed into a 500 mL volumetric flask and filled up with distilled water. |
| Magnesium acetate solution |
w(Mg(CH3COO)2) = 3%
15 g magnesium acetate is weighed into a 500 mL volumetric flask and filled up with glacial acetic acid. |
| Reaction solution |
c(ICl) = 0.1 mol/L in glacial acetic acid
If possible this solution should be bought from a supplier. |
Standard
| . |
. |
| Iodate standard |
Potassium iodate is dried in a drying oven for 2 h at 110 °C and allowed to cool down in a desiccator for at least 1 h. |
Sample Preparation
No sample preparation required.
Analysis
Titer
Around 70 mg potassium iodate is weighed into a 250 mL beaker and then 80 mL distilled water is added to dissolve it. Next, 10 mL β(KI) = 100 g/L, in addition to 25 mL c(H2SO4) = 0.5 mol/L are put into the solution. The solution becomes dark brown and the originated iodine is titrated with c(Na2S2O3) = 0.1 mol/L up to the first end point.
Blank
25 mL c(ICl) = 0.1 mol/L, 20 mL glacial acetic acid, and 10 mL w(Mg(CH3COO)2) = 3% are placed into a 250 mL brown glass beaker. The beaker is left standing for five minutes after being closed with a lid. 15 mL β(KI) = 100 g/L is placed into the solution and the originated iodine is titrated with c(Na2S2O3) = 0.1 mol/L until the first end point.
Sample
An appropriate amount of sample is weighed into a 250 mL brown glass beaker (see table below) and put onto the sample rack. Next, between 20 and 25 mL of glacial acetic acid (see below), 25 mL c(ICl) = 0.1 mol/L and 10 mL w(Mg(CH3COO)2) = 3% are then added. The beaker is then closed with a lid and left to stand for five minutes. 15 mL β(KI) = 100 g/L is placed in the solution and the originated iodine is titrated with c(Na2S2O3) = 0.1 mol/L until the first end point.
Table 3: Sample sizes dependent on the expected iodine value.
| Expected IV / g / 100 g |
Sample amount / g |
Solvent volume / mL |
| < 1.5 |
15.00 |
25 |
| 1.5 – 2.5 |
10.00 |
25 |
| 2.5 – 5 |
3.00 |
20 |
| 5 – 20 |
1.00 |
20 |
| 20 – 50 |
0.40 |
20 |
| 50 – 100 |
0.20 |
20 |
| 100 – 150 |
0.13 |
20 |
| 150 – 200 |
0.10 |
20 |
Parameters
Titer
| . |
. |
| Mode |
DET U |
| Pause |
20 s |
| Signal drift |
20 mV/min |
| Max. waiting time |
38 s |
| Meas. point density |
4 |
| Min. increment |
50 μL |
| Max. increment |
off |
| EP criterion |
5 |
| EP recognition |
greatest |
Blank/Sample
| . |
. |
| Mode |
DET U |
| Signal drift |
20 mV/min |
| Max. waiting time |
38 s |
| Meas. point density |
4 |
| Min. increment |
10 μL |
| Max. increment |
off |
| EP criterion |
5 |
| EP recognition |
all |
Calculations

Example Determination
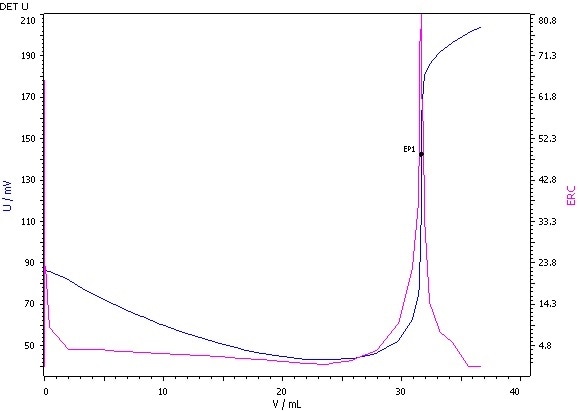
Figure 2: Determination of the iodine value (blue = titration curve, pink = ERC).
Comments
- The technique for determining the iodine value is adapted from AOAC 993.20, AOCS Cd 1d-92, EN ISO 3961, ASTM D5554, USP<401> Method II, and Ph.Eur. 2.5.4 Method B. The following modifications are made:
- Glacial acetic acid is utilized as a solvent instead of a mixture of cyclohexane and glacial acetic acid.
- Magnesium acetate is utilized as catalyst, shortening the reaction time from 1 – 2 hours to 5 minutes.
- The technique is different from AOAC 920.159 in the following ways:
- Instead of tetrachloromethane, glacial acetic acid is utilized as a solvent.
- Magnesium acetate is utilized as catalyst, shortening the reaction time from 0.5 – 1 hour to 5 minutes.
References
- EN ISO 3961 Animal and vegetable fats and oils – determination of iodine value
- ASTM D5554 Standard test method for determination of the iodine value of fats and oils
- AOAC 920.159 Iodine Absorption Number of Oils and Fats
- AOAC 993.20 Iodine Value of Fats and Oils
- AOCS Cd 1d-92 Sampling and analysis of commercial fats and oils: iodine value of fats and oils cyclohexane-acetic acid method
- USP<401> Fats and fixed oils
- Ph.Eur. 2.5.4 Iodine value
Peroxide Value
Summary
The peroxide number supplies information about the amount of peroxide compounds present in the oil and so can be utilized as an indicator of the quality and age of the edible oil. The lower the peroxide numbers the better and/or fresher the oil.
Instruments
- Sample changer with Swing Head and DIS-Cover
- Titrator with DET mode
- 1x Burette 5 mL
- 1x Burette 10 mL
- 3x Burette 20 mL
- 2x Burette 50 mL
- Propeller Stirrer
Electrodes
| . |
. |
| iPt Titrode |
6.0471.300 |
Reagents
- Sulfuric acid, c(H2SO4) = 0.5 mol/L, volumetric solution
- Potassium iodate, KIO3, p.a.
- Potassium iodide, KI, p.a.
- Sodium thiosulfate, c(Na2S2O3) = 0. 1 mol/L, volumetric solution
- Glacial acetic acid, p.a.
- 1-Decanol, p.a.
Solutions
| . |
. |
| Titrant |
c(Na2S2O3) = 0.01 mol/L or 0.001 mol/L
Prepared by dilution of the c(Na2S2O3) = 0.1 mol/L with distilled water. |
| Auxiliary solution |
Saturated solution of KI. |
| Potassium iodide solution |
w(KI) = 10%
50 g potassium iodide is weighed into a 500 mL volumetric flask and filled up with distilled water. |
| Solvent mixture |
Glacial acetic acid / 1-decanol with approximately 20 mg I2 / L
Φ(1-decanol) = 40% (v/v) |
Standard Solution
| . |
. |
| Iodate standard |
Potassium iodate is dried in a drying oven for 2 h at 110 °C and allowed to cool down in a desiccator for at least 1 h.
Approximately 0.65 g is weighed into a 1 L volumetric flask and filled up to the mark with dist. water. |
Sample Preparation
No sample preparation is required.
Analysis
Titer
Between 0.75 to 1.25 mL potassium iodate standard solution is dosed into a 250 mL beaker. 25 mL c(H2SO4) = 0.5 mol/L, 80 mL distilled water, in addition to 10 mL w(KI) = 10%, are added to the solution. The solution becomes dark brown and the originated iodine is titrated with c(Na2S2O3) = 0.01 mol/L or c(Na2S2O3) = 0.001 mol/L until the first end point.
Blank
0.5 mL auxiliary solution and 50 mL solvent mixture are put into a 250 mL brown glass beaker and closed with the DIScover. 80 mL distilled water is added after a minute, and the solution is titrated with c(Na2S2O3) = 0.01 mol/L or c(Na2S2O3) = 0.001 mol/L up until the first end point.
Sample
Depending on the expected value, 5 or 10 g of sample is weighed into a 250 mL brown glass beaker and put onto the sample rack. 0.5 mL auxiliary solution and 50 mL solvent mixture are added and the beaker is closed with the lid of the OMNIS DIS-cover system. 80 mL distilled water is added after one minute, and the solution is titrated with c(Na2S2O3) = 0.01 mol/L or c(Na2S2O3) = 0.001 mol/L until the first end point.
Parameters
Titer
| . |
. |
| Mode |
DET U |
| Pause |
20 s |
| Signal drift |
20 mV/min |
| Max. waiting time |
38 s |
| Meas. point density |
4 |
| Min. increment |
50 μL |
| Max. increment |
500 μL |
| EP criterion |
5 |
| EP recognition |
all |
Blank/Sample
| . |
. |
| Mode |
DET U |
| Signal drift |
5 mV/min |
| Min. waiting time |
10 s |
| Max. waiting time |
72 s |
| Meas. point density |
4 |
| Min. increment |
10 μL |
| Max. increment |
200 μL |
| EP criterion |
20 |
| EP recognition |
greatest |
Calculations
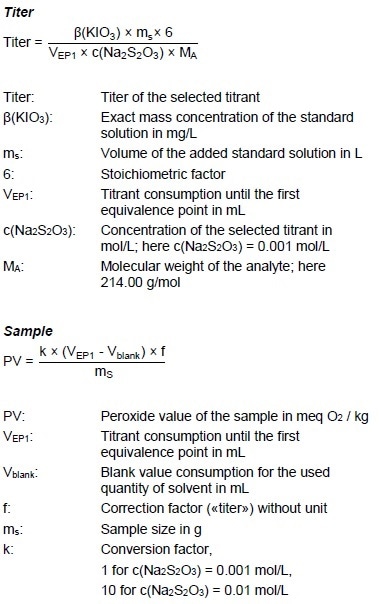
Example Determination
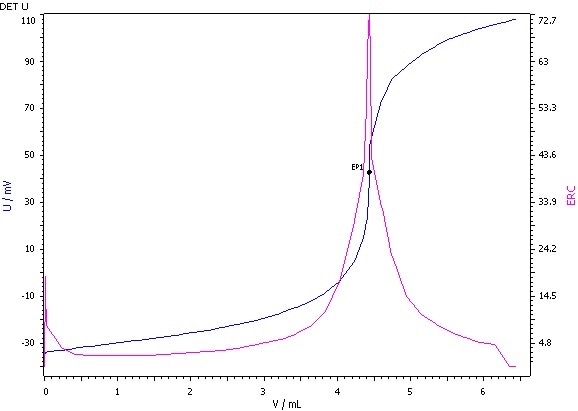
Figure 3: Determination of the peroxide value (blue = titration curve, pink = ERC).
Comments
- All dosing units, particularly the one for the solvent mixture, must be prepared prior to each determination series. This ensures that, during a series, the amount of iodine added together with the solvent mixture is always the same.
- Samples with peroxide numbers which are less than 1 are titrated with c(Na2S2O3) = 0.001 mol/L as a higher consumption of titrant leads to better reproducibility.
- For dissolving of the oil, the stirrer must be set to a higher speed (14), than for the titration (10), or results may not be reproducible.
- To allow the determination of reliable blank value, a small amount of iodine is added to the solvent mixture. However, a condition for this is that exactly the same amount (10.0 mL) of solvent mixture be utilized for each calculation of both the blank and the sample.
- It was decided by ISO/TC 34/SC 11 to keep the sample size to fixed to 5 g for PV greater than 1, and to 10 g for PV less than or equal to 1, as the peroxide value is dependent on the sample size.
- The technique differs from AOAC 965.33, USP<401> and Ph.Eur. 2.5.5 Method A in the following way:
- A mixture of 1-decanol and glacial acetic acid is employed as solvent instead of a mixture of chloroform and glacial acetic acid.
- 50 mL of solvent is used instead of 30 mL.
- The technique for determining the peroxide value is adapted from EN ISO 27107 and EN ISO 3960. The following alterations are made:
- H2SO4 instead of HCl is utilized in the titer determination.
- A mixture of 1-decanol and glacial acetic acid is utilized as a solvent rather than a mixture of isooctane and glacial acetic acid.
References
- EN ISO 27107 Animal and vegetable fats and oils – determination of peroxide value – potentiometric end-point determination
- EN ISO 3960 Animal and vegetable fats and oils -- Determination of peroxide value -- Iodometric (visual) endpoint determination
- AOAC 965.33 Peroxide Value of Oils and Fats
- USP<401> Fats and fixed oils
- Ph.Eur. 2.5.5 Peroxide value
Saponification Value
Summary
The saponification number is employed as a quality control parameter and for the characterization of oils and fats. The saponification number carries the information on the average molecular weight of all fatty acids which are present in the sample. The higher the saponification number, the lower the molecular weight of all fatty acids, and vice versa.
Instruments
- Titrator with DET mode
- Burette 50 mL
- Stirrer
- Reflux condenser
- Heating device
Electrodes
| Solvotrode easyClean |
6.0229.020 |
Reagents
- Hydrochloric acid, c(HCl) = 0.5 mol/L, volumetric solution
- Potassium hydroxide, p.a.
- Ethanol, p.a.
- TRIS, p.a.
Solutions
| . |
. |
| Titrant |
c(HCl) = 0.5 mol/L
If possible this solution should be bought from a supplier. |
| Ethanolic potassium hydroxide solution |
c(KOH) = 0.5 mol/L in ethanol
If possible this solution should be bought from a supplier.
The solution should be colorless or straw yellow. For the preparation of a stable colorless solution see paragraph 5.1 of ISO 3657. |
| Electrolyte |
c(TEABR) = 0.4 mol/L in ethylene glycol
Metrohm No. 6.2320.000 |
Standard
| . |
. |
| TRIS |
TRIS is dried over night in a drying oven at 105 °C and allowed to cool down in a desiccator for at least 1 h. |
Sample Preparation
Weigh out an appropriate amount of sample in a round-bottomed flask (see table below). Add a magnetic stirring bar and 25 mL of ethanolic c(KOH) = 0.5 mol/L. Next, attach the reflux cooler, heat up and boil gently for an hour, intermittently tilting the flask back and forth.
Table 4: Sample sizes dependent on the expected saponification value.
| Expected SV / mg KOH / g |
Sample amount / g |
| 150 – 200 |
2.2 – 1.8 |
| 200 – 250 |
1.7 – 1.4 |
| 250 – 300 |
1.3 – 1.2 |
| > 300 |
1.1 – 1.0 |
Analysis
Titer
About 420 mg TRIS is weighed into a titration vessel. 50 mL ethanol and 20 mL deionized water are added. The solution is titrated with c(HCl) = 0.5 mol/L to the first equivalence point after a pause of 20 seconds. The electrode membrane is rehydrated for 1 minute in deionized water between measurements.
Blank
For the blank test, carry out a sample preparation without sample. Dilute the flask contents sufficiently with ethanol after cooling, insert the electrode and the burette tip, then backtitrate the KOH excess with c(HCI) = 0.5 mol/L up to the first equivalence point. Rehydrate the electrode membrane for 1 minute in deionized water between measurements.
Sample
Dilute the flask contents sufficiently with ethanol after cooling, insert the electrode and the burette tip, then backtitrate the KOH excess with c(HCI) = 0.5 mol/L to the first equivalence point. Rehydrate the electrode membrane for 1 minute in deionized water between measurements.
Parameters
Titer
| . |
. |
| Mode |
DET U |
| Pause |
20 s |
| Signal drift |
20 mV/min |
| Max. waiting time |
38 s |
| Meas. point density |
4 |
| Min. increment |
50 μL |
| Max. increment |
off |
| EP criterion |
5 |
| EP recognition |
greatest |
Blank/Sample
| . |
. |
| Mode |
DET U |
| Pause |
20 s |
| Signal drift |
20 mV/min |
| Max. waiting time |
38 s |
| Meas. point density |
4 |
| Min. increment |
10 μL |
| Max. increment |
off |
| EP criterion |
5 |
| EP recognition |
greatest |
Calculations

Example Determination
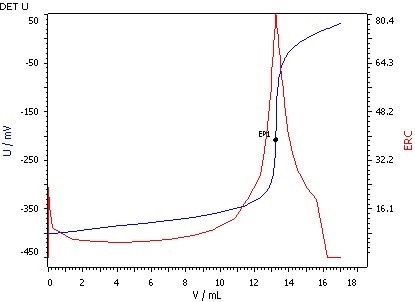
Figure 4: Determination of the saponification value (blue = titration curve, red = ERC).
Comments
- The potassium hydroxide solution should be straw yellow or colorless. A description for the preparation of a stable colorless solution is located in the norm ISO 3657.
- Samples which are challenging to saponify should be boiled for 2 hours.
- Ph.Eur. 2.5.6 differs from the technique outlined herein that they utilize different sample sizes depending on the expected saponification value.
- AOAC 920.160 and ASTM D5558 differ from the technique outlined here, in that they use 50 mL of KOH solution and so, also a higher sample size of between 4 and 5 g.
- For more information about the handling of the Solvotrode easyClean, see the instructions sent with the electrode.
References
- EN ISO 3657 Animal and vegetable fats and oils – determination of saponification value
- AOAC 920.160 Saponification Number (Koettstorfer Number) of Oils and Fats
- ASTM D5558 Standard Test Method for Determination of the Saponification Value of Fats and Oils
- USP<401> Fats and fixed oils
- Ph.Eur. 2.5.6 Saponification value
Acid value, Free Fatty Acids
Summary
The acid value or acid number, and the free fatty acid content are employed for the determination of oils and fats and as quality control parameter. The higher the acid number and free fatty acid content, the lower the quality of the oil. The acid number increases further with the age of an oil, because triglycerides are changed into glycerol and fatty acids as an effect of time.
Instruments
- Sample changer
- Titrator with DET mode
- Burette 20 mL
- Stirrer
Electrodes
| . |
. |
| Solvotrode easyClean |
6.0229.020 |
Reagents
- Ethanol, p.a.
- Diethyl ether, peroxide-free, p.a.
- Phenolphthalein, p.a.
Solutions
| . |
. |
| Titrant |
c(KOH) = 0.1 mol/L in ethanol or methanol
If possible this solution should be bought from a supplier. |
| Solvent mixture |
Ethanol / diethyl ether, Φ(EtOH) = 50% (v/v)
Neutralized, just before use, with KOH in presence of 0.3 mL phenolphthalein solution per 100 mL solvent mixture. |
| Phenolphthalein solution |
Phenolphthalein in ethanol, β(phenolphthalein) = 1 g / 100 mL. |
Standard
| . |
. |
| Benzoic acid |
Benzoic acid is dried in a desiccator over night. |
Sample Preparation
No sample preparation required.
Analysis
Titer
100 – 120 mg benzoic acid is weighed into the titration vessel and dissolved in 50 mL ethanol. Next, the solution is titrated using c(KOH) = 0.1 mol/L until after the first equivalence point.
Sample
An appropriate sample volume is placed into a 150 mL beaker (see table below). Next, 50 to 100 mL solvent mixture is added and the sample dissolved. The solution is titrated after a pause of 30 seconds until the first equivalence point, by utilizing alcoholic c(KOH) = 0.1 mol/L.
Table 5: Sample sizes dependent on the expected acid value.
| Expected AV / mg KOH / g |
Sample amount / g |
Accuracy / g |
| 0 – 1 |
20 |
0.05 |
| 1 – 4 |
10 |
0.02 |
| 4 – 15 |
2.5 |
0.01 |
| 15 - 75 |
0.5 |
0.001 |
| > 75 |
0.2 |
0.001 |
Parameters
Titer
| . |
. |
| Mode |
DET U |
| Signal drift |
50 mV/min |
| Max. waiting time |
26 s |
| Meas. point density |
4 |
| Min. increment |
10 μL |
| Max. increment |
off |
| EP criterion |
5 |
| EP recognition |
all |
Sample
| . |
. |
| Mode |
DET U |
| Signal drift |
20 mV/min |
| Max. waiting time |
38 s |
| Meas. point density |
4 |
| Min. increment |
10 μL |
| Max. increment |
off |
| EP criterion |
5 |
| EP recognition |
all |
Calculations

Example Determination
Table 6: Choice of fatty acids for the free fatty acid content
| Type of fat |
Expressed as |
Molar mass / g/mol |
| Coconut oil, Palm kernel oil and similar oils |
Lauric acid |
200 |
| Palm oil |
Palmitic acid |
256 |
| Oils from certain cruciferae |
Erucic acid |
338 |
| All other fats |
Oleic acid |
282 |
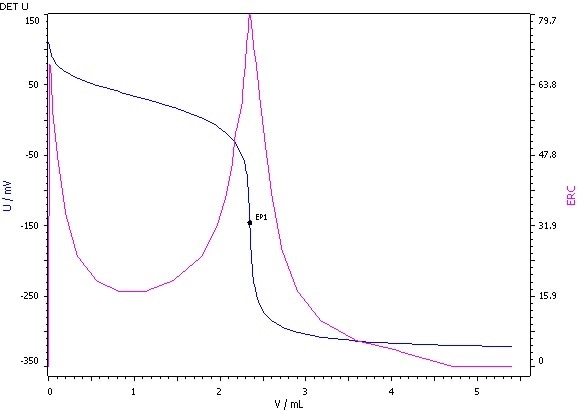
Figure 5: Determination of the acid value (blue = titration curve, pink = ERC).
Comments
- In the instance of rapeseed oil, which possesses a maximum of erucic acid content of 5%, the acidity shall be given as oleic acid.
- If the results of the free fatty acids are simply reported as acidity, without further definition, this is expressed as oleic acid by convention. If the sample contains mineral acids, these are established as fatty acids by convention.
- For animal or hard fats, a solvent mixture of one volume ethanol and three volumes tert-butyl methyl ether or toluene is advised. This mixture should also be neutralized.
- Ph.Eur 2.5.1 differs from the technique described here in that petrol ether 100-120 °C is employed in the solvent mixture instead of diethyl ether.
- For more information about the handling of the Solvotrode easyClean, please see the leaflet sent with the electrode.
References
- EN ISO 660 Animal and vegetable fats and oils – determination of acid value and acidity
- USP<401> Fats and fixed oils
- Ph.Eur. 2.5.1 Acid value
- Application Bulletin AB-315 Determination of free fatty acids (FFA) in edible oils with 859 Titrotherm
Hydroxyl Value
Summary
For establishing the hydroxyl value, most standards require long refluxing times and/or toxic pyridine. The determination described below, according to ASTM E1899 has the benefit that neither long refluxing times nor toxic pyridine are needed.
Instruments
- Sample changer
- Titrator with DET mode
- 1x Burette 50 mL (acetonitrile)
- 2x Burette 20 mL (reaction solution, titrant)
- 1x Burette 2 mL (distilled water)
- Magnetic stirrer for sample changer
- DIS-Cover
Electrodes
| . |
. |
| Solvotrode easyClean |
6.0229.010 |
Reagents
- Acetonitrile, HPLC quality
- Toluene-4-sulfonyl-isocyanate, purum (TSI)
- Ethanol, purity >99.8 %
- Potassium hydrogen phthalate, KHP, p.a.
Solutions
| . |
. |
| Titrant |
Tetrabutyl ammonium hydroxide, c(TBAOH) = 0.1 mol/L in isopropanol/methanol, Φ(MeOH) = 50% (v/v)
If possible, this solution should be bought from a supplier. |
| TSI solution |
The solution reacts vigorous with water, it is therefore recommended to work in a fume hood and under protective gas.
Approximately 250 mL acetonitrile is given into a 500 mL volumetric flask and 20 mL TSI is added. The flask is filled up to the mark with acetonitrile and mixed.
The reaction solution is stable for approximately 1 month. |
Standard
| . |
. |
| KHP |
KHP is dried in a drying oven for 2 h at 120 °C and allowed to cool down in a desiccator for at least 1 h. |
Sample Preparation
No sample preparation required.
Analysis
Titer
60 mL distilled water is added to around 180 mg KHP and the suspension stirred for approximately a minute to dissolve the KHP. Then, the solution is titrated until the first equivalence point using c(TBAOH) = 0.1 mol/L.
Sample
An appropriate amount of sample (see the below calculation) is put into the titration vessel and dissolved in 10 mL acetonitrile. Next, the beakers are covered and the solution is stirred for 30 seconds using stirring rate 8. 10.0 mL TSI solution are added and the sample is covered again and the mixture stirred using stirring rate 4. 0.5 mL distilled water is added after 5 minutes, the lid is closed again and the solution is stirred for another minute using stirring rate 4.
40 mL acetonitrile is added and then the solution is titrated until after the second end point with c(TBAOH) = 0.1 mol/L. The burette and vessel are rinsed with ethanol first, then with distilled water and the electrode is conditioned for 1 minute in distilled water after every titration.

Parameters
Titer
| . |
. |
| Mode |
DET U |
| Pause |
30 s |
| Signal drift |
50 mV/min |
| Max. waiting time |
26 s |
| Meas. point density |
4 |
| Min. increment |
10 μL |
| Max. increment |
off |
| EP criterion |
5 |
| EP recognition |
greatest |
Sample
| . |
. |
| Mode |
DET U |
| Pause |
30 s |
| Signal drift |
50 mV/min |
| Max. waiting time |
26 s |
| Meas. point density |
4 |
| Min. increment |
10 μL |
| Max. increment |
off |
| EP criterion |
5 |
| EP recognition |
all |
Calculations

Example Determination
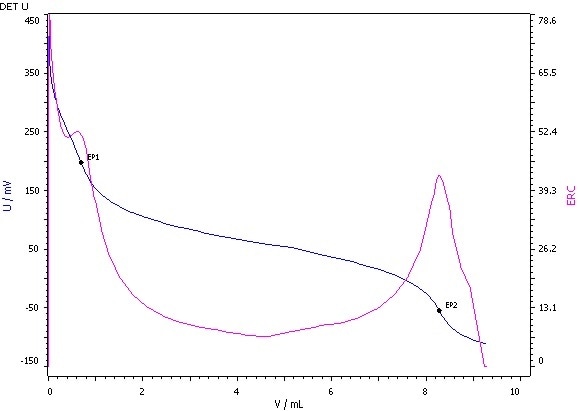
Figure 6: Determination of the hydroxyl value (blue = titration curve, pink = ERC).
Comments
- The ASTM technique is shown here, as it is quicker (12 min) than technique EN ISO 4629-2 (40 minutes).
- For more information regarding the handling of the Solvotrode easyClean, see the leaflet sent with the electrode.
References
- ASTM E1899-08 Standard test method for hydroxyl groups using reaction with p-toluene sulfonyl isocyante (TSI) and potentiometric titration with tetrabutyl ammonium hydroxide
- Application Bulletin AB-322 Fully automated potentiometric determination of the hydroxyl number (HN) according to ASTM E1899 and EN ISO 4629-2
Nickel Traces
Summary
The production of margarine often involves the hardening of liquid oils by a catalytic hydrogenation of the fatty acids, nickel is one of the catalysts employed for this process. Polarography can be employed to identify traces of nickel impurities in the final product.
Instruments
- VA instrument capable of operating a mercury electrode and supporting DP mode
Electrodes
| . |
. |
. |
| WE |
Multi-Mode Electrode pro (MME pro)
Mercury drop capillary |
6.1246.120
6.1226.030 |
| AE |
Separate Pt rod electrode |
6.0343.000 |
| RE |
Ag/AgCl reference electrode c(KCl) = 3 mol/L
Electrolyte vessel filled with c(KCl) = 3 mol/L |
6.0728.020
6.1245.010 |
Reagents
- Hydrochloric acid, w(HCl) = 30%, for trace analysis*, CAS 7647-01-0
- Ammonium hydroxide solution, w(NH3) = 25%, for trace analysis*, CAS 1336-21-6
- Dimethylglyoxim disodium salt octahydrate, Na2DMG, for analysis, CAS 75006-64-3
- Ni standard stock solution, β(Cu2+) = 1 g/L, commercially available
- Nitric acid, w(HNO3) = 65%, for trace analysis* CAS 7697-37-2
- Ultrapure water, resistivity >18 MΩ·cm (25 °C), type I grade (ASTM D1193)
* e.g. Merck suprapur®, Sigma-Aldrich TraceSelect® or equivalent
Solutions
| . |
. |
| DMG solution |
c(Na2DMG) = 0.1 mol/L
Dissolve 0.304 g Na2DMG in 10 mL ultrapure water. This solution needs to be prepared daily. |
Standard Solution
| . |
. |
| Ni standard |
β(Ni2+) = 10 mg/L
0.5 mL Ni standard stock solution (1 g/L) and 0.05 mL nitric acid (65%) are made up to 50 mL with ultrapure water. |
Sample Preparation
Accurately weigh out a 2.5 g sample in a round-bottomed flask. Add 2.5 mL w(HCl) = 30%, attach a reflux condenser, heat up the solution and boil for 15 minutes. With a small quantity of ultrapure water, rinse out the warm solution into a separating funnel. Separate and gather the aqueous phase.
Using hot ultrapure water, extract the round-bottomed flask and the fatty phase a further three times. Filter the combined aqueous extracts through a paper filter (e.g ‘White Ribbon Filter’ grade 589/2) into a 100 mL volumetric flask, add 5 mL w(NH3) = 25% and fill up to the mark with ultrapure water.
Analysis
Measuring Solution
20 mL sample extract (after sample preparation)
0.1 mL DMG solution.
Pipette 20 mL of the prepared sample solution (corresponding to a 0.5 g portion of the original sample) into the polarography vessel and then add 0.1 mL DMG solution. The pH of the measuring solution must be be 9.5 ± 0.1. The concentration of Ni is quantified by two additions of Ni standard solution β(Ni2+) = 10 mg/L.
Parameters
Volumes
| . |
. |
| Sample amount |
0.5 g |
| Cell volume |
20.1 mL |
Voltammetric
| . |
. |
| Electrode |
DME |
| Mode |
DP – Differential pulse |
| Initial purge time |
300 s |
| Hydrodynamic measurement |
No |
| Sweep |
|
| Start potential |
-0.8 V |
| End potential |
-1.4 V |
| Pulse amplitude |
0.05 V |
| Pulse time |
0.04 s |
| Voltage step |
0.006 V |
| Voltage step time |
0.6 s |
| Sweep rate |
0.01 V/s |
Substance and calibration
| . |
. |
| Name |
Nickel |
| Peak potential |
-1.0 V |
| Tolerance |
0.05 V |
| Calibration method |
Standard addition |
Example Determination
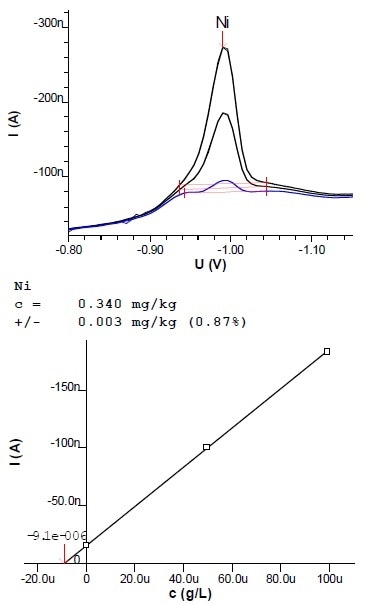
Figure 7: Voltammogram and calibration curve of a determination of Ni in margarine (2.7 g sample extracted into 100 mL)
Comments
- To establish the reagent blank, the sample preparation procedure is performed as described, only without the sample. The blank concentration is established with the same parameters as for the sample. The blank concentration is then taken away from the sample concentration.
- Combustion as decomposition is not viable as volatile nickel carbonyl is lost in the process.
About Metrohm
At Metrohm is one of the world’s most trusted manufacturers of high-precision instruments for chemical analysis. Metrohm was founded in 1943 by engineer Bertold Suhner in Herisau, Switzerland. Today, Metrohm is represented in 120 countries by subsidiaries and exclusive distributors. The global Metrohm Group also includes the Dutch companies Metrohm Applikon and Metrohm Autolab, manufacturers of online analyzers and instruments for electrochemical research, respectively. Recently, the Metrohm Group was joined by Metrohm Raman, a leading manufacturer of handheld Raman spectrometers.
Metrohm is the global market leader in analytical instruments for titration. Instruments for ion chromatography, voltammetry, conductivity, and stability measurement make the Metrohm portfolio for ion analysis complete. Instruments for Near-infrared and Raman spectroscopy are another, strongly growing segment of the Metrohm portfolio.
Metrohm is a problem solver, both in the laboratory and within the industrial process. To this end, the company offers their customers complete solutions, including dedicated analytical instrumentation as well as comprehensive application know-how. More than 30% of the company’s employees at the Metrohm international headquarters in Herisau work in R&D.
Metrohm has been owned 100% by the non-profit Metrohm Foundation since 1982. The Metrohm Foundation, which does not exert any influence on the company’s business operations, sponsors gifted students in the natural sciences, supports charitable and philanthropic purposes and, above all, ensures the independence of the company.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.