Jun 28 2018
Researchers at Juntendo University report in the journal Leukemia how mutants of the protein calreticulin lead to molecular mechanisms triggering myeloproliferative neoplasms, which can cause cancer. The findings may lead to the development of novel therapies for certain types of blood cancer.
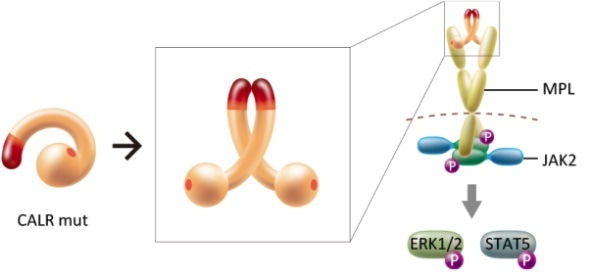
The formation of multimers of mutant CALR is found to play a key role in the pathogenesis of CALR-mutant myeloproliferative neoplasms. The structural change induced by the homomultimerization makes MPL bind to the multimer complex, which in turn activates a particular signaling pathway.
Myeloproliferative neoplasms (MPNs) are a class of diseases in which the body produces too many blood cells or platelets (thrombocytes) in the bone marrow. MPNs can lead to cancers, including acute myeloid leukemia. It has been established that in some patients with MPNs, mutation of calreticulin (CALR) occurs; CALR is a protein capable of binding to other, misfolded proteins, triggering their degradation. Mutant CALR, in turn, is known to promote the activation of another protein, thrombopoietin receptor, associated with the development of cancers. Now, a team of researchers led by Marito Araki from Juntendo University has understood the mechanism behind the activation of thrombopoietin receptor through mutant CALR. Their findings have important consequences for developing new therapies against tumors induced by mutant CALR.
The researchers started from the hypothesis that the interaction between mutant CALRs mediates a particular association between two thrombopoietin receptor molecules (also known as MPL molecules, the abbreviation referring to myeloproliferative leukemia). This assumption was based on earlier work by the research team establishing that mutant CALR indeed activates MPL and a set of signaling molecules, one of which is known as JAK2, triggering cancer.
Marito Araki et al., Blood 127:1307-1316, (2016)
http://www.bloodjournal.org/content/127/10/1307?sso-checked=true
A key observation made by the scientists is that mutant CALR molecules form homomultimeric complexes (clusters of identical proteins), whereas non-mutant (‘wild-type’) CALRs do not. Araki and colleagues could attribute the multimerization to particular chemical motifs in the mutant molecules. The researchers then confirmed that the formation of homomultimeric complexes indeed leads to the activation of MPL, by comparing with experiments performed on wild-type CALR.
Based on their observations, the scientists propose that the actual activation happens through homomultimeric mutant CALR interacting simultaneously with two MPL molecules, which in turn triggers the formation of JAK2 and subsequent biochemical signalling pathways during MPN development. The researchers conclude that their insights show that inhibiting the intermolecular interaction provides a way to prevent tumor formation, but point out that further studies are needed:
Although more detailed molecular and structural analyses are required to understand the mechanism behind MPL activation by mutant CALR proteins, our findings shed light on MPN pathogenesis and provide support for the development of novel therapeutic strategies against MPNs with mutant CALR proteins."
Source: https://www.juntendo.ac.jp/english/
 SickKids study uncovers cell recovery mechanisms linked to stomach cancer
SickKids study uncovers cell recovery mechanisms linked to stomach cancer