An interview with Mingje Xie, CEO of Rapid Novor, conducted by James Ives
Please give an overview of W-ion based Isoleucine Leucine Determination (WILD™) technology and how it helps to ensure 100% accuracy in antibody protein sequencing.
The purpose of antibody protein sequencing is to accurately deduce every single amino acid present in the primary sequence. Antibody proteins expressed from primary codes incorrectly sequenced may elicit drastically different binding behaviour when compared to the original antibody. Even a single amino acid error in the primary sequence may have a damaging impact on the final antibody structure.
._3D_illustration-Tatiana_Shepeleva.jpg)
Shutterstock | Tatiana Shepeleva
Out of the 20 amino acids, isoleucine (Ile) and leucine (Leu) have the same molecular weight and cannot be distinguished from one another using conventional mass spectrometry (MS) experiments. Protein sequencing via conventional MS methods therefore lacks reliable accuracy.
To solve this problem, we developed WILD™. WILD™ stands for W-ion Isoleucine Leucine Determination technology and takes advantage of the structural differences between Ile and Leu. WILD™ fully utilizes advanced fragmentation technology, such as electron transfer high-energy collision dissociation (EThcD), built into the MS instrument, to break the side chains (orange scissors in Figure 1) in order to produce satellite w-ions. The w-ions produced during the fragmentation will have different mass shifts, which can accurately and reliably identify the specific residues.

Figure 1. Representation of W-ion Isoleucine Leucine Determination (WILDTM). The orange scissors pinpoint where the sidechains are broken to release satellite w-ions and accurately and reliably distinguish between isoleucine and leucine.
Why is it important to be able to differentiate Leucine and Isoleucine especially in CDRs?
The complementarity determining region (CDR) of antibodies has a highly specific sequence for each antigen. Therefore, the order and type of residue present in the CDR is crucial for the function of the antibody.
Though isobaric in weight, Ile and Leu have different structures that dictate different antibody-antigen interactions. We have observed that Ile and Leu structural differences can affect the antibody's binding affinity, and in some cases, when incorrect Ile or Leu residues are in or around CDR regions, binding no longer occurs.
How prevalent are Leucine and Isoleucine in antibody proteins and in particular in CDRs?
That’s an interest question because the number of Ile and Leu in the CDRs can be quite random. Figure 2 is a modified version from a poster we published at the annual meeting for the American Society for Mass Spectrometry in 2018.
The poster depicts data generated from several hundreds of monoclonal antibodies we sequenced. As you can see, some antibodies have no Isoleucine or Leucine in any of the six CDRs. However, on the other extreme, we observed some cases with a total of nine Ile or Leu in the CDRs. On average, however, CDRs will contain at least three to five Ile or Leu residues.
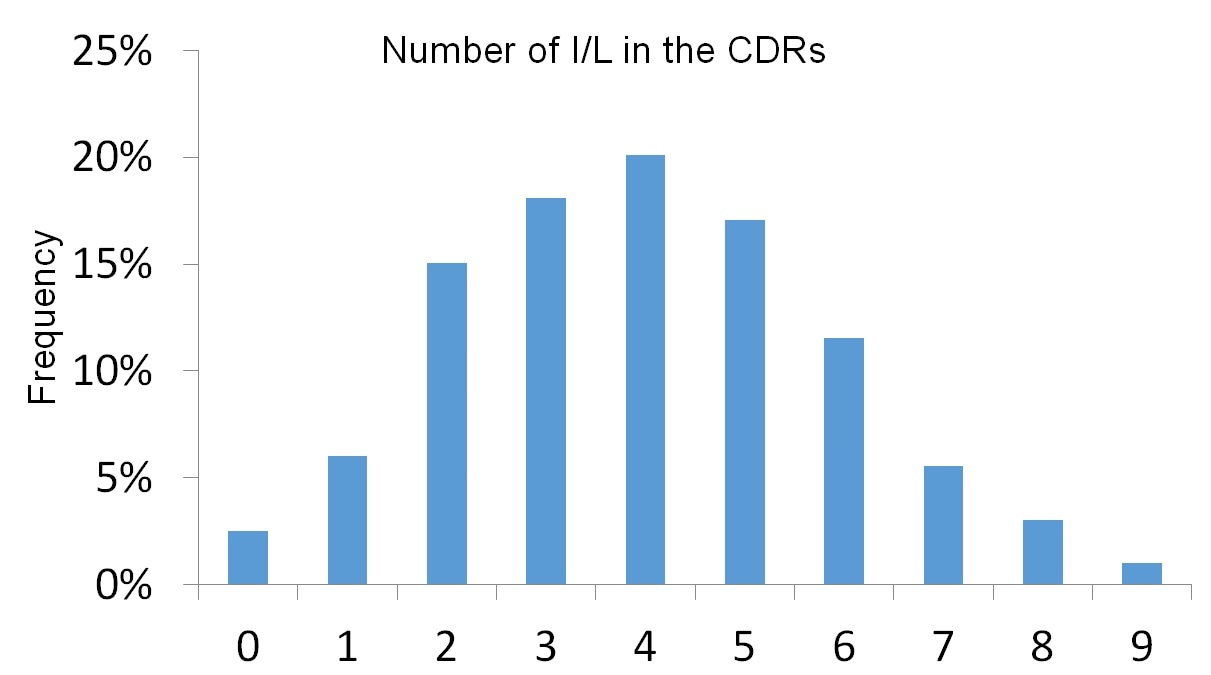
Figure 2. Frequency of antibodies expressing a given number of Isoleucine (I) or Leucine (L) residues in the complementarity determining region (CDR).
Without determining the difference between leucine and isoleucine, how would a researcher determine that an antibody works in practice?
Without an accurate determination of the two isobaric amino acids, researchers can only use permutation to predict all possible Ile or Leu positions within the sequence. However, this might be neither feasible nor economical in many cases.
For example, for projects where antibodies have nine Ile or Leu, it might not be even practical for the researcher to generate expressing all 500 or more possible combinations in order to identify the correct, let alone functional, antibodies.
Until recently what was the gold standard in this type of sequencing? Please give an overview of these techniques and their strengths and weaknesses when compared to WILD™.
Edman degradation was the most commonly used technique for sequencing proteins. This technique relies on a cyclical process that involves chemical catalysis to orchestrate the repeated removal of amino acids starting from the N-terminus of the peptide, and then the detection of the amino acid via electrophoresis, chromatography and mass spectrometry.
Edman degradation has considerable drawbacks. Firstly, the process requires a large amount of starting material. Secondly, because the process relies on labelling the N-terminus prior to removal, it is unable to process peptides with inaccessible N-termini.; Thirdly, Edman sequencing throughput is very low. One cycle of the Edman degradation could take up to one hour.
Finally, in practice, Edman degradation can only sequence about 30 amino acids. Ile and Leu-containing peptides would need to be isolated from the mixture of peptides via electrophoresis or chromatography prior to identifying all Ile and Leu residues because conventional MS experiments cannot distinguish Ile from Leu. This task is cumbersome and inefficient. It would take days to sequence all the Ile and Leu containing peptides. Though Edman sequencing was very important for elucidating the sequence of antibodies, this technology has many pitfalls and is essentially outdated nowadays.
Most recently, fragmentation of proteins into peptides has been achieved through enzymatic catalysis followed by MS analysis. Cleavage sites on the antibody protein of interest are initially identified to predict potential fragments, and then compared with the observed fragments using MS experiments.
Commonly, the enzyme chymotrypsin is used to generate a digest pattern that can help distinguish Ile from Leu residues. We also have employed enzymatic digestion of antibodies in the past. We noted that enzymes purchased from different vendors, used at different conditions, on different antibodies, behaved differently. We also found that the enzymatic error rate was very high and that the method was often irreproducible and unreliable. We thus sought to develop a high accuracy, repeatability and reliability method: WILD™.
How to sequence antibody proteins using mass spec?
What are the limitations of WILD™ technology?
Our WILD™ technology is still fairly new, both in terms of the technology development and the market awareness. However, we continue to increase its throughput and accuracy. Currently, WILDTM can determine a sample sequence with 90% accuracy in a single round; we employ other rounds to ensure repeatability and reach 100% accuracy.
This need for additional rounds with complex mixtures translates to delays, but with continuous improvement, we hope to drastically decrease throughput time. We continue to perform carefully designed experiments for complex antibodies, so that in the future we can determine all Ile and Leu residues in a sample with 100% accuracy. We are particularly invested and eager to embark in determining the sequence of antibodies in polyclonal and oligoclonal mixtures.
What is the future for WILD™ technology and Rapid Novor?
We hope that WILDTM technology can be used to guide future recombinant efforts to guide vaccine rational design and immunotherapeutic strategies.
Where can readers find more information?
Our website, https://www.rapidnovor.com/resources/wild/, is a great resource for information related to WILD™. You can also find more information related to antibody protein sequencing, at this URL https://www.rapidnovor.com/antibody/.
About Mingjie Xie
Mr. Mingjie Xie, MSc, MBA, is the co-founder and CEO of Rapid Novor Inc. He is a computer scientist by training, received his MSc degree from Western University in the field of bioinformatics. He received his MBA degree from Richard Ivey School of Business to pursue his interests in business. Prior to co-founding Rapid Novor Inc, Mingjie was the COO of a bioinformatics software company.
 Rapid Novor receives Ontario Ministry of Health provisional license for EasyM® blood-based MRD test for multiple myeloma
Rapid Novor receives Ontario Ministry of Health provisional license for EasyM® blood-based MRD test for multiple myeloma