All circulating variants of concern (VOCs) of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) contain mutations in the spike (S) glycoprotein, specifically within its receptor-binding domain (RBD). Several previous studies have attempted to understand the effect of RBD mutations in circulating VOCs, primarily focusing on the RBD- angiotensin-converting enzyme 2 (ACE2) interaction.
The formation of the RBD-ACE2 complex is the first step in the entry of SARS-CoV-2 into a host cell, which involves the fusion of viral S protein, via its RBD, with the ACE2 receptor on host cell membranes. Although several studies have explored the mechanistic implications and pharmaceutical potential of the SARS-CoV-2 RBD-ACE2 complex, these studies have not given much attention to the conformational dynamics of the RBD.
Interestingly, upon viral attachment, host factors trigger large-scale conformational rearrangements in the S protein that lead to membrane fusion and viral entry.
About the study
Many naturally occurring mutations in the SARS-CoV-2 S protein have arisen in the RBD region. Some of these mutations have given significant fitness advantages to SARS-CoV-2 variants by increasing the RBD-ACE2 binding affinity or facilitating escape from anti-SARS-CoV-2 monoclonal antibodies. To date, there is limited data on the impact of these mutations on the RBD’s conformational dynamics.
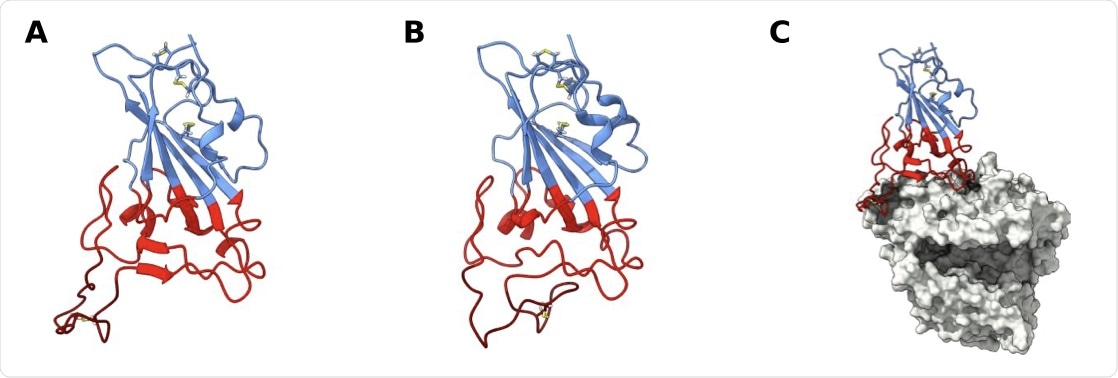 SARS-CoV-2 receptor binding domain (RBD) structure. Structure of wt RBD in the open (A) and closed (B) confor- mations. Snapshots obtained from the AA MD simulations. Disulfide bonds are represented in yellow sticks. Structure of wt RBD bound to ACE2 is also shown (C). The RBM region is colored red and the ridge in dark red, with the rest of the protein being colored in blue. ACE2 is in grey.
SARS-CoV-2 receptor binding domain (RBD) structure. Structure of wt RBD in the open (A) and closed (B) confor- mations. Snapshots obtained from the AA MD simulations. Disulfide bonds are represented in yellow sticks. Structure of wt RBD bound to ACE2 is also shown (C). The RBM region is colored red and the ridge in dark red, with the rest of the protein being colored in blue. ACE2 is in grey.
In a recent study published on the bioRxiv* preprint server, a team of researchers investigate the conformational dynamics of the SARS-CoV-2 RBD using the atomistic (AA) molecular dynamics (MD) simulation method. The researchers also characterize the impact of mutations on RBD conformational dynamics in solution through the use of the principal component analysis (PCA) method, which reduced coordinates from the simulations to two principal components or two-dimensional (2D) configuration space that were expressed as free energy landscapes.
Study findings
Visual inspection of the wild-type (wt) RBD in an aqueous solution revealed that large dynamic conformational changes occur in the receptor-binding motif (RBM) region, appearing as an opening and closing of the ACE2 binding surface of the RBM.
The study results showed that the wt RBD adopted two distinct conformations in equilibrium including an open conformation, where the RBD freely bonded with ACE2, and a closed conformation, where the RBM ridge blocked the ACE2 binding surface and hindered binding to ACE2. Upon characterizing these two states, it was observed that they originated from specific intramolecular interactions between residues of the RBM ridge and those of the ACE2 binding surface.
According to the authors, this “hinge-like” mechanism that can effectively shield the ACE2 binding surface from the solvent and binding partners was reported for the first time. Although this mechanism has yet to be observed in experimentally-solved RBD structures, the researchers believe that the RBM is found unresolved in most structures due to the flexibility of the region. Those that are fully resolved are structures of RBD complexed with either ACE2, antibodies, or dimerized by the ACE2 binding surface.
It was observed that the SARS-CoV-2 Alpha and Beta variants significantly shifted the equilibrium of wt RBD towards more open conformations by roughly 20%. This shift towards more open conformations must have enhanced the binding affinity of ACE2 by increasing accessibility to the RBM and facilitating binding.
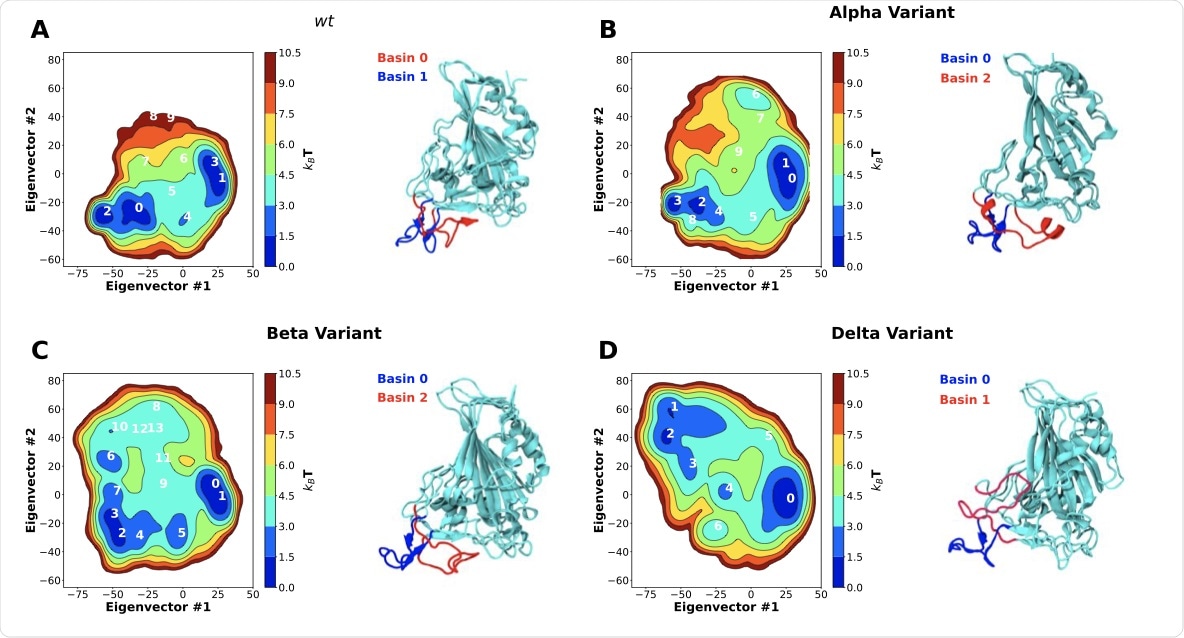 Two-dimensional principal component analysis (PCA) of SARS-CoV-2 RBD conformational dynamics in water. Plots of the first two principal components determined from the C-alpha backbone of the wt RBD (A) as well as the alpha (B), beta (C) and delta (D) variants. Snapshots of the lowest energy structures for selected basins are also shown. Basins were numbered according to their free energy.
Two-dimensional principal component analysis (PCA) of SARS-CoV-2 RBD conformational dynamics in water. Plots of the first two principal components determined from the C-alpha backbone of the wt RBD (A) as well as the alpha (B), beta (C) and delta (D) variants. Snapshots of the lowest energy structures for selected basins are also shown. Basins were numbered according to their free energy.
Comparatively, the Delta variant showed a completely different alternative open conformation that was unseen in the other variants. In this conformation, the ACE2 binding surface remained open and accessible for binding; however, there was a significant alteration in the conformation of the RBM ridge, which resulted in backward bending towards the RBD core, shielding a part of it from exposure. The researchers hypothesized that this may provide a fitness advantage to the Delta variant by aiding in antibody escape, as many RBD targeting antibodies are known to target the RBM ridge region.
Recently, a new VOC denoted as the B.1.1.529 (Omicron) variant has been detected, which contains 15 mutations, 10 of which are located in the RBM region. Some of these mutations like K417N, T478K, E484K, and N501Y were similar to the mutations observed in the Alpha, Beta, and Delta variants. Based on the observations of this study, these mutations would have heavily impacted the open/closed equilibrium for wt RBD, much like the alternative conformations observed in the Delta variant, thus improving its antibody escape and providing the Omicron variant substantial fitness advantage.
To summarize, the findings of the current study indicate that the mutations found in the Alpha, Beta, and Delta variants impact RBD conformational dynamics in such a way that stimulates efficient binding to ACE2 and offers antibody escape capabilities to these variants.
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
Source: Valero, M. F., Borges-Araujo, L., Melo, M. N., et al. (2021). SARS-CoV-2 variants impact RBD conformational dynamics and ACE2 accessibility. bioRxiv. doi:10.1101/2021.11.30.470470. https://www.biorxiv.org/content/10.1101/2021.11.30.470470v2.
- Peer reviewed and published scientific report.
Valério, Mariana, Luís Borges-Araújo, Manuel N. Melo, Diana Lousa, and Cláudio M. Soares. 2022. “SARS-CoV-2 Variants Impact RBD Conformational Dynamics and ACE2 Accessibility.” Frontiers in Medical Technology 4 (October). https://doi.org/10.3389/fmedt.2022.1009451. https://www.frontiersin.org/articles/10.3389/fmedt.2022.1009451.
 Early immune responses linked to broadly neutralizing antibodies against HIV
Early immune responses linked to broadly neutralizing antibodies against HIV