The emergence of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) triggered the ongoing coronavirus disease 2019 (COVID-19). Both immunologically and biologically, the SARS-CoV-2 spike protein represents an attractive target for researchers looking to counteract or neutralize this virus.
Study: A pseudovirus system enables deep mutational scanning of the full SARS-CoV-2 spike. Image Credit: Design_Cells / Shutterstock.com
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Introduction
DMS is a valuable tool for predicting how thousands of distinct genetic changes in the spike protein can affect viral behavior mediated by the effects on protein folding, protein function, and antibody binding. As a high-throughput method, DMS can generate large amounts of data ideal for computational modeling methods to predict the antigenic behavior of potential new SARS-CoV-2 variants.
Until the current study, DMS was primarily used to explore mutations on the receptor binding domain (RBD) of the spike protein, or a small number of mutations on the spike, using cell surface display systems on yeast or mammalian cells. However, this approach limits the ability of DMS to measure neutralization effects accurately; therefore, only antibody binding results are assessed, which are often less clinically relevant.
A novel DMS platform
The main novelty of the current platform is its unique protocol designed to generate large numbers of pseudovirus mutants in libraries, with each lentivirus genotype linked to a specific surface spike protein variant. Such genotype-phenotype linkage is not typical in such experiments.
While the absence of such a linkage does not pose a problem when using a single viral variant, the current experiment sought to study thousands of variants. To this end, the new platform was used to map the effects of mutations in different parts of the RBD, N-terminal domain (NTD), and S2 subunit of the spike protein on cell infection and antibody neutralization.
The approach used in the current study can generate over 105 transduction units (genotype-phenotype linked pseudoviruses) per milliliter, with a further increase by five- to ten-fold possible in the right conditions.
The researchers selected only mutations that seemed probable during natural viral evolution. These mutations were obtained come from the Global Initiative for Sharing Avian Influenza Data (GISAID) database, thus likely representing the functional spike protein.
Mutations that have independently emerged multiple times were used at a higher rate, whereas sites exhibiting positive selection were intensively interrogated by including every possible change in amino acids. Frequently observed deletions were also included.
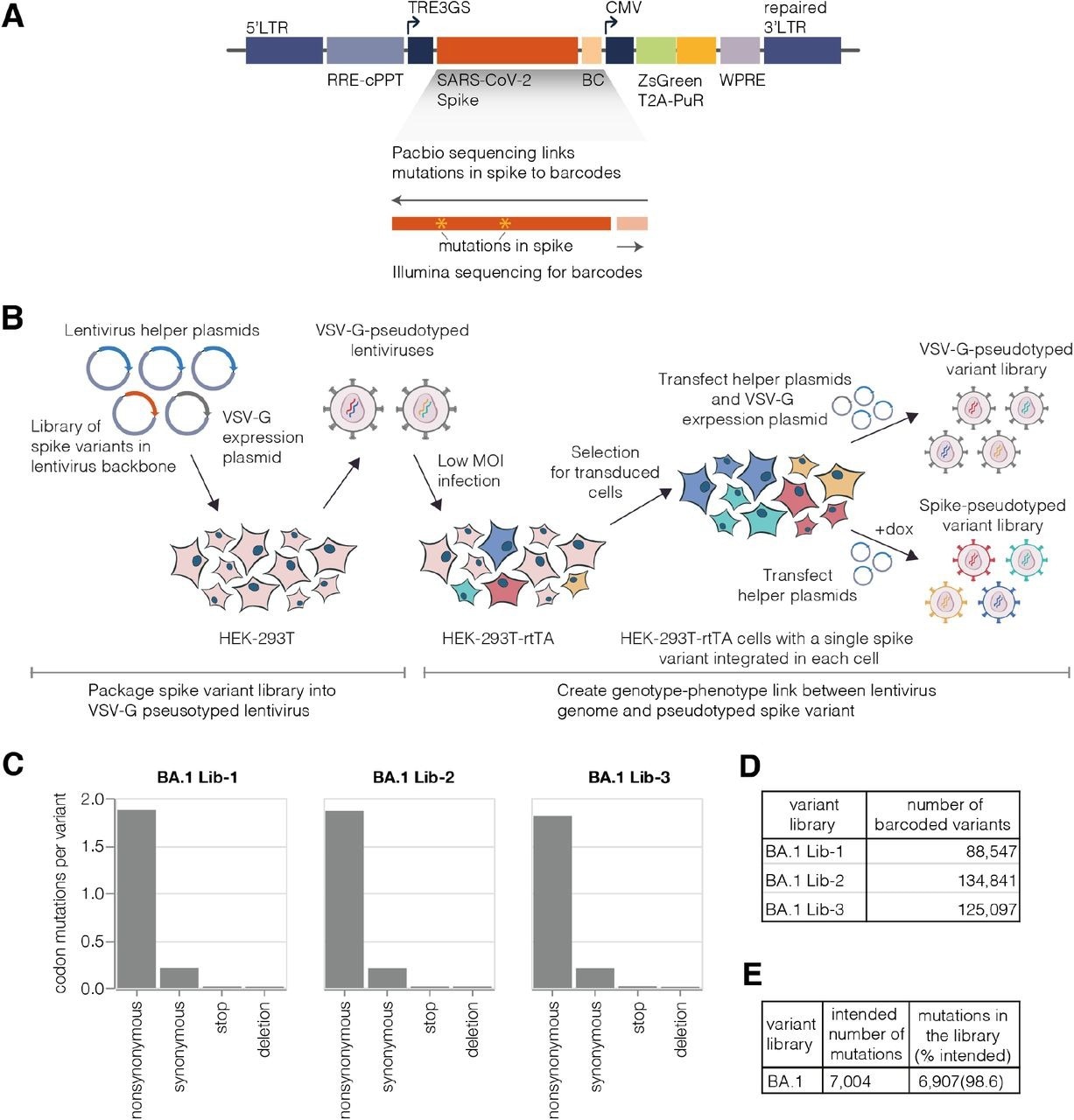 Deep mutational scanning platform for spike. (A) Lentivirus backbone used for deep mutational scanning. The backbone contains functional lentiviral 5’ and 3’ long terminal repeat (LTR) regions. The spike gene is under an inducible tet response element 3rd generation (TRE3G) promoter, and there is a 16 nucleotide barcode (BC) downstream of the stop codon. A CMV promoter drives expression of reporter ZsGreen gene that is linked to a puromycin resistance gene (PuR) via a T2A linker. The backbone also contains a woodchuck hepatitis virus posttranscriptional regulatory element (WPRE), Rev response element (RRE), and a central polypurine tract (cPPT). (B) Approach for creating genotype-phenotype linked lentivirus libraries. HEK-293T cells are transfected with spike-carrying lentivirus backbone, VSV-G expression plasmid and lentiviral helper plasmids to generate VSV-G-pseudotyped lentiviruses. These viruses are used to transduce reverse tetracycline-controlled transactivator (rtTA) expressing HEK-293T cells at low multiplicity of infection (MOI), and successfully transduced cells are selected using puromycin. Selected cells can be transfected with helper plasmids and a VSV-G expression plasmid to produce VSV-G-pseudotyped viruses carrying all genomes present in the deep mutational library or selected cells can be induced with doxycycline (dox) to express spike and transfected with only the helper plasmids to generate spike-pseudotyped lentiviruses that have a genotype-phenotype link. (C)Average number of mutations per barcoded spike in BA.1 libraries. (D) Total number of barcoded variants in each BA.1 library. (E) The coverage of intended mutations across all BA.1 libraries.
Deep mutational scanning platform for spike. (A) Lentivirus backbone used for deep mutational scanning. The backbone contains functional lentiviral 5’ and 3’ long terminal repeat (LTR) regions. The spike gene is under an inducible tet response element 3rd generation (TRE3G) promoter, and there is a 16 nucleotide barcode (BC) downstream of the stop codon. A CMV promoter drives expression of reporter ZsGreen gene that is linked to a puromycin resistance gene (PuR) via a T2A linker. The backbone also contains a woodchuck hepatitis virus posttranscriptional regulatory element (WPRE), Rev response element (RRE), and a central polypurine tract (cPPT). (B) Approach for creating genotype-phenotype linked lentivirus libraries. HEK-293T cells are transfected with spike-carrying lentivirus backbone, VSV-G expression plasmid and lentiviral helper plasmids to generate VSV-G-pseudotyped lentiviruses. These viruses are used to transduce reverse tetracycline-controlled transactivator (rtTA) expressing HEK-293T cells at low multiplicity of infection (MOI), and successfully transduced cells are selected using puromycin. Selected cells can be transfected with helper plasmids and a VSV-G expression plasmid to produce VSV-G-pseudotyped viruses carrying all genomes present in the deep mutational library or selected cells can be induced with doxycycline (dox) to express spike and transfected with only the helper plasmids to generate spike-pseudotyped lentiviruses that have a genotype-phenotype link. (C)Average number of mutations per barcoded spike in BA.1 libraries. (D) Total number of barcoded variants in each BA.1 library. (E) The coverage of intended mutations across all BA.1 libraries.
A library of 7,000 mutations in the Omicron BA.1 variant spike and almost the same number of Delta variant spike mutations were used for the current study. The current method used two or three codon mutations in each spike variant to be transduced, thus allowing the effect of each mutation to be assessed against the background of other accompanying mutations.
Each of these mutations received its own barcode after integrating the pseudovirus into its final host cell.
Importantly, linking barcodes to spike variants allows us to use short-read Illumina sequencing of the barcode to obtain the full spike genotype in all subsequent experiments.”
With the genotype-phenotype protocol, two and three libraries of Delta and BA.1 variants were created, containing 50,000 and 100,000 variants each, respectively. These libraries were assigned functional scores reflecting their ability to infect a host cell relative to the parental spike variant. Non-synonymous mutations often had poor scores, especially when multiple, “reflecting the cost of accumulating multiple often mildly deleterious mutations.”
The researchers also added a barcoded pseudovirus with a spike variant resistant to all anti-spike antibodies. This provided an absolute neutralization standard in which changes in pseudovirus barcode counts for this and other variants could be compared to calculate the degree of change in neutralization for each variant relative to this variant.
What did the study show?
A test case using a set of BA.1 neutralizing antibodies (nAbs) targeting the RBD (LY-CoV1404 or bebtelovimab), NTD (5-7), and S2 (CC67.105) was then conducted.
Bebtelovimab remained potent against Omicron because of its RBD epitope. While some Omicron mutations at escape sites in the bebtelovimab epitope have little effect, others escaped. As new K444 mutations are being detected in newer Omicron subvariants, bebtelovimab may eventually lose its existing potency.
Similar results were obtained with the other two antibodies as well, in which sites, where complete escape can be produced were identified.
The breadth of neutralization, that is, the ability to neutralize pan-sarbecovirus epitopes, does not confer potency against SARS-CoV-2. This is due to the presence of mutations detected in some Omicron subvariants that also allow complete escape from broadly neutralizing antibodies.
[This] emphasizes the importance of directly mapping escape mutations in SARS-CoV-2 in addition to assessing breadth across other natural SARS-related coronaviruses.”
The effect of various mutations on viral infection in the absence of antibodies was also explored. To this end, the platform could identify different deleterious and beneficial effects for various mutations. The researchers also examined how functional effects could reflect the actual evolution of the virus.
Interestingly, the results were accurate when compared with real-world observations as compared to earlier experiments, “presumably because our experiments mimic the true biological function of spike better than cell-surface display experiments.” Nevertheless, mutations with positive functional scores were not enriched during natural evolution, perhaps due to the difference in selection pressures between the laboratory and natural environments.
Early D614G mutations remain unsurpassed in their ability to improve infectivity.
Conclusions
Taken together, the study findings provide highly accurate large-scale measurements of the impact of spike mutations. Thus, the platform discussed here could help identify the most likely potential future escape mutations.
The novel platform described in the current study could be invaluable in assessing direct escape from neutralization in polyclonal sera, which often affects antibody binding and neutralization differently. Furthermore, this method can be used to assess new spike variants and thus direct rational vaccine design.
The researchers also indicate that their platform provides new directions for therapeutic antibody development by revealing how broadly neutralizing antibodies remain susceptible to immune escape mutations in SARS-CoV-2, thus emphasizing the importance of conserved spike regions.
The link between genotype and viral phenotype is vital in understanding how natural selection pressures affect the evolution of the spike protein. Importantly, this platform can be used for any virus that can be pseudotyped for antigenic surveillance and vaccine design.
Given the potential to introduce gain-of-function into potential pandemic pathogens, such experiments must be regulated appropriately to avoid creating new risks while advancing public health benefits.
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
Dadonaite, B., Crawford, K. H. D., Radford, C. E., et al. (2022). A pseudovirus system enables deep mutational scanning of the full SARS-CoV-2 spike. bioRxiv. doi:10.1101/2022.10.13.512056. https://www.biorxiv.org/content/10.1101/2022.10.13.512056v1.
- Peer reviewed and published scientific report.
Dadonaite, Bernadeta, Katharine H. D. Crawford, Caelan E. Radford, Ariana G. Farrell, Timothy C. Yu, William W. Hannon, Panpan Zhou, et al. 2023. “A Pseudovirus System Enables Deep Mutational Scanning of the Full SARS-CoV-2 Spike.” Cell 186 (6): 1263-1278.e20. https://doi.org/10.1016/j.cell.2023.02.001. https://www.cell.com/cell/fulltext/S0092-8674(23)00103-4.
 Scientists develop a dual action strategy to destabilize the COVID virus
Scientists develop a dual action strategy to destabilize the COVID virus