 *Important notice: medRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
*Important notice: medRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
Background
Despite extensive scientific investigations conducted throughout the pandemic, the pathophysiology of COVID-19 largely remains elusive. Although SSCTA has discerned the immune signatures of the microenvironment for severe COVID-19, such as diffuse alveolar damage (DAD) that manifests heterogeneously within patients and across infected tissues, analysis of these complex datasets remains challenging.
About the study
In the present study, researchers used five COVID-19 autopsies and one uninfected postmortem case to retrieve tissues for SSCTA analysis. All tissues exhibited pulmonary thromboembolism, lymphocytic infiltration, and DAD of varying degrees after staining with hematoxylin and eosin (H&E).
Using in situ sequencing, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genome and 221 cellular genes induced by its infection were identified. Lung tissues were then stained with 4′,6-diamidino-2-phenylindole (DAPI) for cell segmentation.
SSCTA data was extensively studied using a self-developed workflow, which led to the identification of 869,453 to 3,424,675 reads in the six lung tissues. Then, the Baysor algorithm was applied to segment these reads into cells, which led to the detection of 186,659 to 470,294 cells for each tissue sample, which were further filtered to obtain high-quality cells.
SSCTA also allowed for the identification of the local impact of SARS-CoV-2 infection on lung pathology. The local infection rate analysis through Moran’s I statistics was then applied.
Local feature distributions in the neighboring region of the cell with a fixed cell number or radius were also calculated. This approach allowed for the generation of spatial gene expression and co-expression maps of ligand-receptor pairs.
Uniform Manifold Approximation and Projection (UMAP) was used to visualize the relationship of individual cell gene expressions associated with different cell types in all tissues. Next, individual cells were mapped to their spatial locations in the tissues for neighborhood cell type composition (NCTC) analysis.
Results
The study workflow allowed for the identification of 10,414,863 deoxyribonucleic acid (DNA) transcripts in lung tissues from five COVID-19 patients and one normal lung tissue. This workflow appeared robust and scalable for targeted empirical analyses of lung tissues emphasizing spatial discovery.
Transcript-based cell segmentation detected 93% of DNA transcripts in 1,719,459 cells, of which only a subgroup of 779,137 high-quality cells was isolated. Cell typing these cells with marker genes showed that SARS-CoV-2 infected 18 cell types in the lungs; however, less than 5% of these cells were infected in the tissue samples.
Differential expression analysis of genes in SARS-CoV-2-infected and uninfected lung tissue cells compared to cells in the non-COVID-19 lung tissue revealed that SARS-CoV-2 exerted similar effects on these cells across all tissues. These indirect effects of inflammation and complement activation might also be responsible for lung pathology in COVID-19 patients.
Lung parenchymal cells encompassing alveolar cells (ACs), fibroblasts, and vascular endothelial cells (VECs) were most frequently infected by SARS-CoV-2. COVID-19 lung tissues had fewer ACs and smooth muscle cells (SMCs) but more epithelial cells (ECs) accompanied by infiltrations of innate immune cells, including multiple myeloma (MMs), natural killer (NK) cells, granulocytes, and T and B adaptive immune cells.
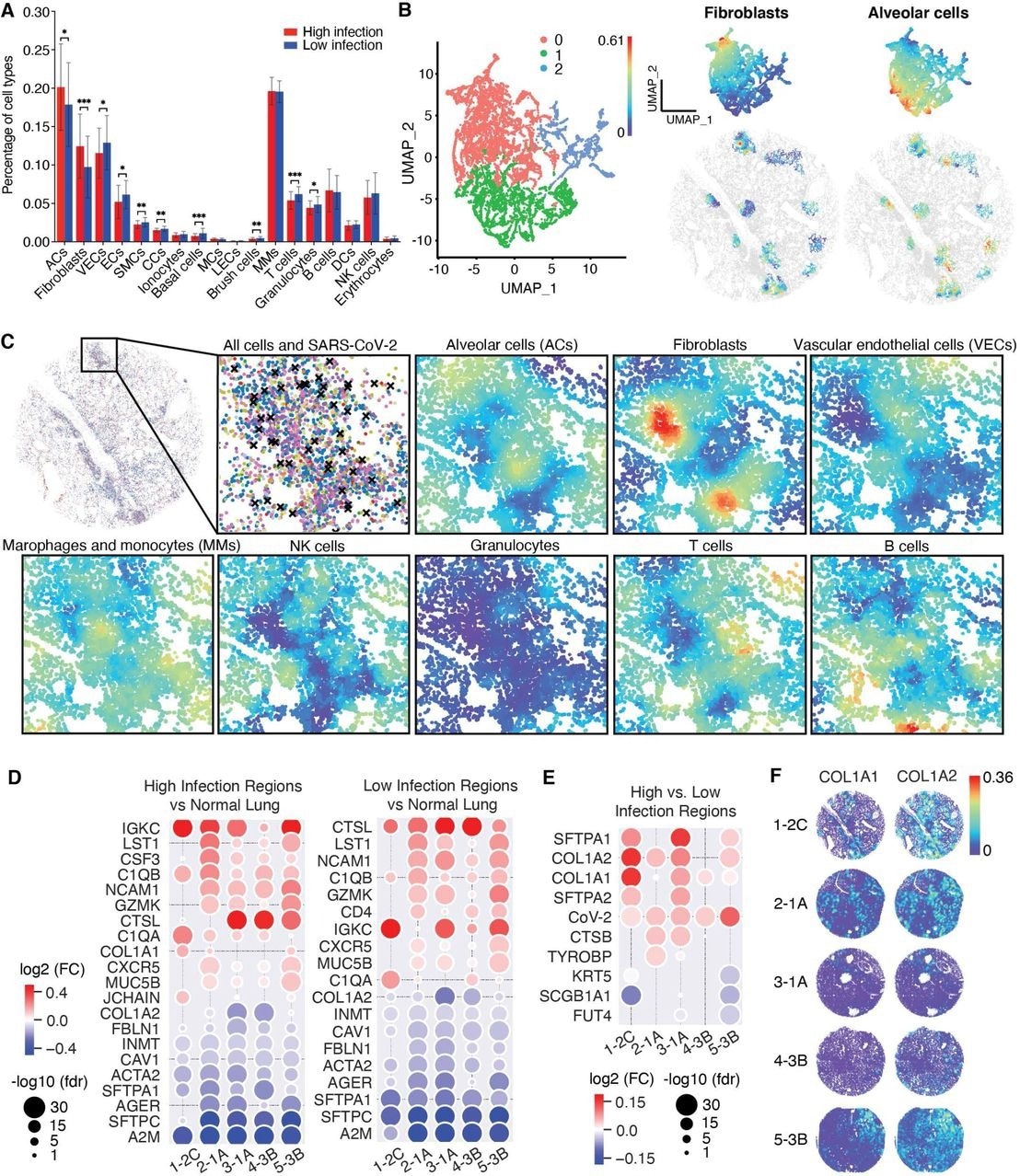 Spatial analysis reveals cellular and gene expression features in regions with high SARS-CoV-2 infection rates. A. A bar plot showing the cell type composition of all identified cell types in the high and low infection regions. The increase in alveolar cells (ACs) might suggest the proliferation of ACs due to injury repair of alveoli and alveoli capillaries and a decrease in epithelial cells (ECs) and vascular endothelial cells (VECs) might suggest the damages caused by the infection. B. UMAP projection of the NCTC analysis results of high infection regions (refer to figure 3B) revealed three distinct clusters, of which two were distinctly enriched in local compositions of ACs and fibroblasts, illustrated on the right with their spatial visualizations. C. Spatial visualization of the compositions in high infection regions revealed negative correlation of fibroblasts and ACs (-0.6, Pearson correlation), which overlapped with annotated OP regions (high fibroblasts, low ACs). D. Differential expressions of high and low infection regions of the COVID-19 tissues compared to the non-COVID-19 tissue, which shares 18 of the 20 differentially expressed genes, confirming the strong indirect effect of SARS-CoV-2 infection. E. Differential expressions between the high and low infection regions of the COVID-19 tissues revealed distinct differentially expressed genes showing upregulation of AC markers SFTPA1 and SFTPA2 in tissues 1, 3, and 5, and fibroblast markers COL1A1 or COL1A2 in all COVID-19 tissues. F.Spatial visualization of the spatial gene expression map of COL1A1 and COL1A2 markers.
Spatial analysis reveals cellular and gene expression features in regions with high SARS-CoV-2 infection rates. A. A bar plot showing the cell type composition of all identified cell types in the high and low infection regions. The increase in alveolar cells (ACs) might suggest the proliferation of ACs due to injury repair of alveoli and alveoli capillaries and a decrease in epithelial cells (ECs) and vascular endothelial cells (VECs) might suggest the damages caused by the infection. B. UMAP projection of the NCTC analysis results of high infection regions (refer to figure 3B) revealed three distinct clusters, of which two were distinctly enriched in local compositions of ACs and fibroblasts, illustrated on the right with their spatial visualizations. C. Spatial visualization of the compositions in high infection regions revealed negative correlation of fibroblasts and ACs (-0.6, Pearson correlation), which overlapped with annotated OP regions (high fibroblasts, low ACs). D. Differential expressions of high and low infection regions of the COVID-19 tissues compared to the non-COVID-19 tissue, which shares 18 of the 20 differentially expressed genes, confirming the strong indirect effect of SARS-CoV-2 infection. E. Differential expressions between the high and low infection regions of the COVID-19 tissues revealed distinct differentially expressed genes showing upregulation of AC markers SFTPA1 and SFTPA2 in tissues 1, 3, and 5, and fibroblast markers COL1A1 or COL1A2 in all COVID-19 tissues. F.Spatial visualization of the spatial gene expression map of COL1A1 and COL1A2 markers.
UMAP helped allow for the visualization of separated clusters for ACs and fibroblasts. However, immune cells remained grouped and mixed with VECs.
Consistent with pathological investigations, NCTC analysis revealed unorganized distributions of ACs, VECs, and fibroblasts, which were the major parenchymal cell types in SARS-CoV-2-infected tissues. In addition, differential gene expression evaluations of COVID-19 and non-COVID-19 lung tissues identified nine and ten up and downregulated genes, respectively.
Only three of the 19 genes were differentially expressed genes (DEGs), which suggests that cytokines and complement activation induced by SARS-CoV-2 infection likely contribute to the dysregulated gene expressions and pathology in COVID-19 lung tissues.
A positive spatial correlation was observed between cell densities and SARS-CoV-2 infection rates, thus suggesting that increased cell densities might be associated with high infection rates. These regions highly expressed SARS-CoV-2 entry-related factors, including angiotensin-converting enzyme 2 (ACE2) and FURIN.
Segmenting regions with high infection rates and cell densities confirmed that the former caused persistent damage to the lung tissues, thereby resulting in the infiltration of immune cells, wound repair biomarkers, and fibrosis.
NCTC analysis showed three clusters in high-infection regions, two of which were distinctly enriched with prominent local compositions of fibroblasts and ACs, respectively. The high and low infection regions sharing 18 of 20 DEGs presented highly consistent differential expression patterns, thus indicating the pronounced indirect effects of SARS-CoV-2 infection.
One of the two differentially expressed genes in high infection regions was cytokine colony-stimulating factor 3 (CSF3), an interleukin-6 (IL-6) cytokine family member, upregulated in all infected tissues. Furthermore, investigating spatial co-expression of CSF3 and its receptor revealed that their co-expression also exhibited spatial correlations in high infection regions.
Conclusions
The current study presented an atlas of spatial patterns of different cellular features that characterize SARS-CoV-2 infection and the immune infiltrations, inflammation, and damage induced by severe disease. These results offer crucial insights into the cellular spatial mechanisms that contribute to the development of acute respiratory distress syndrome (ARDS) in COVID-19 patients.
Overall, the study findings demonstrate the power of spatial single-cell transcriptomics in studying COVID-19 lung pathology and other health and disease conditions.
*Important notice: medRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
 ESCRT-III proteins play an unexpected role in protecting DNA bridges during cell division
ESCRT-III proteins play an unexpected role in protecting DNA bridges during cell division