SARS-CoV-2, which is the virus responsible for the coronavirus disease 2019 (COVID-19), has caused the deaths of over 4.4 million lives worldwide, thus making it the greatest pandemic since 1918. To date, there are no specific antivirals available to treat SARS-CoV-2. As a result, the research community has turned to repurposing drugs and developing monoclonal antibodies to fight the virus.
The virus envelope of SARS-CoV-2 is covered with the spike glycoprotein. The spike protein mediates viral engagement with the host cell angiotensin-converting enzyme 2 (ACE2) receptor that ultimately allows for viral entry into the cell.
Most of the current COVID-19 vaccines use the spike antigen to generate antibodies and cellular immunity, including memory cells, to recognize and neutralize the virus. The spike receptor-binding domain (RBD) is among the top targets for many monoclonal antibodies (MAbs), as well as those elicited by natural infection.
MAbs are generated by cloned B-cells that originate from a single parent cell. All MAbs have the same protein sequence and bind to the target epitope with high affinity and antigen specificity.
Convalescent plasma (CP) also transfers specific antibodies to the recipient; however, unlike MAbs, CP carries the risk of blood-borne disease. To date, the two MAbs of Bamlanivimab and REGN-COV2 have been granted emergency use authorization (EUA) by the United States Food and Drug Administration (FDA).
New variants emerge
The emergence of new and potentially resistant variants of SARS-CoV-2 poses a formidable challenge to the efficacy of current MAbs and vaccines. These variants carry multiple mutations in the RBD, such as the N501Y mutation in the alpha (B.1.1.7) variant, which rapidly became the dominant circulating strain worldwide after its identification towards the end of 2020.
N501Y is associated with immune evasion, though it is neutralized by current vaccine-elicited antibodies and MAbs. The beta variant (B.1351) carries K417N and E484K mutation in its RBD and is ten-fold less capable of being neutralized by these antibodies.
Comparatively, the kappa variant B.1.617 carries the RBD mutations L452R and E484Q, while the delta variant B.1.617.2 has 50-67% greater infectivity than the parent strain. The alpha, beta, and delta variants have all been referred to as variants of concern (VOCs), as they resist neutralization, are more infective, or both.
Exploring RBD-ACE2 interactions
The current study investigated the altered biological characteristics of the mutants in terms of the direct and allosteric interactions between the mutant RBD and the ACE2 receptor.
The researchers used molecular dynamics (MD) simulations to understand the dynamic perturbations that occur at the RBD-ACE2 interface. To this end, four RBDs were analyzed in the current study, with one being the wildtype and the others obtained from the alpha, beta, and kappa variants. The authors also used two antibodies of B38 and BD23, which have two or more of the four mutated residues in these RBD variants.
These antibodies were sourced from CP, each with one heavy and one light chain, termed A and B, respectively. The antigen-binding fragment (Fab) of B38 is smaller than that of BD23. Additionally, B38 has RBD residues 417 and 501, whereas the BD23 epitope contains residues at 452, 484, and 501.
About the study
The effect of each mutant RBD on structure, relative to the wildtype virus, was examined. The antigen-antibody complex formed by each mutant was then studied for allosteric interactions.
The mechanism by which the complex dissociates was also explored for each mutant by non-equilibrium pulling simulations and subsequently tested against enthalpy and hydrogen bonding forces.
Study findings
The RBD is a relatively rigid system as a result of the many beta-sheet configurations. Comparatively, antibodies are very flexible due to their large conformational changes, as shown by the high root mean squared deviation (RMSD). However, BD23 is uniformly flexible, which is comparable to B38, in which the distant domain of which is far more flexible than the antigen-binding domain.
The wild-type and mutant RBD-antibody complexes are similar in their overall root mean squared fluctuation (RMSF). At specific residues, flexibility changed markedly in bound and unbound states at residues 452, 484, and 501, denoted by different RMSF.
Both E484Q and N501Y mutations are in the region comprising residues 460-510, where both antibodies bind. The greatest change in flexibility is in the loop formed by residues 470-490, with the kappa and alpha variants showing 2-3 times greater and lower flexibility, respectively. Notably, the flexibility of the loop was highest in the unbound state.
With the B38-Fab complex, the K/Q484 and adjacent residues become twice as flexible as the wildtype E484 residue. Comparably, for the BD23-Fab, the RMSF drops to half. The L452R-E484Q mutant RBD is most rigid in complex and most flexible unbound.
Interestingly, N501Y and the K417N-E484K-N501Y mutants showed the highest overall increase in flexibility in complex with antibodies, but no change in RMSF when unbound.
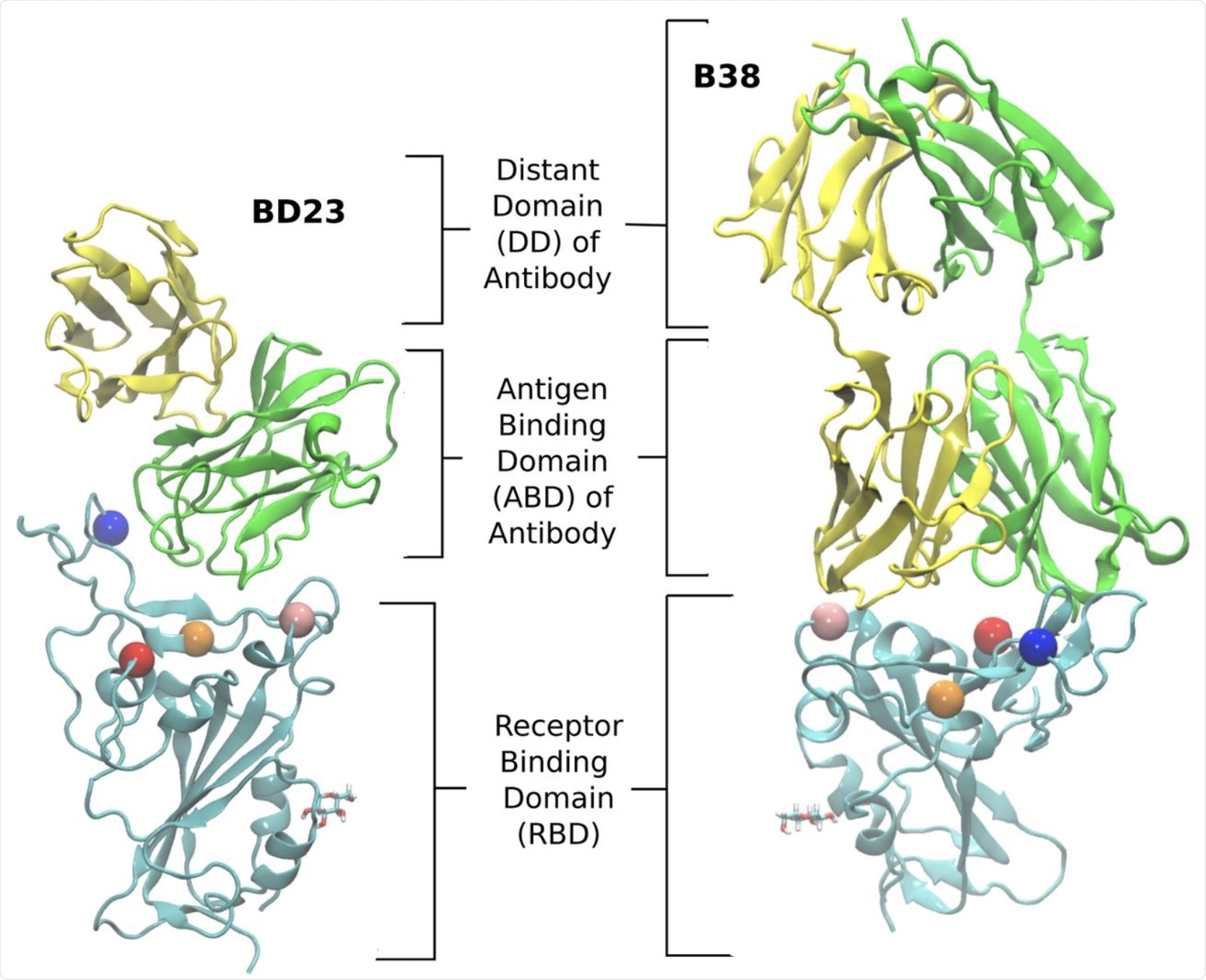 The structure of the RBD-antibody complexes for the two neutralizing antibodies, BD23 and B38-Fab, studied in this work. Each antibody has two chains: the heavy and the light chain. We refer to the heavy chain as chain A and the light chain as chain B. The chain A is colored green and the chain B is colored yellow. The RBD is colored cyan. We define the antigen binding domain (ABD) of the Ab as the domain of the antibody which comes in contact with the RBD. The rest of the antibody is referred to as the distant domain (DD). The Glycan moiety on Asn343 is shown in licorice. The residues which are mutated in the studied variants are represented as colored spheres with the following color scheme: K417 (red), L452 (orange), E484 (blue) and N501 (pink).
The structure of the RBD-antibody complexes for the two neutralizing antibodies, BD23 and B38-Fab, studied in this work. Each antibody has two chains: the heavy and the light chain. We refer to the heavy chain as chain A and the light chain as chain B. The chain A is colored green and the chain B is colored yellow. The RBD is colored cyan. We define the antigen binding domain (ABD) of the Ab as the domain of the antibody which comes in contact with the RBD. The rest of the antibody is referred to as the distant domain (DD). The Glycan moiety on Asn343 is shown in licorice. The residues which are mutated in the studied variants are represented as colored spheres with the following color scheme: K417 (red), L452 (orange), E484 (blue) and N501 (pink).
Antibody RMSF
“It [the antibody RMFS change] serves as a direct proof that secondary perturbations arising from mutations in the antigenic epitope can influence the microscopic dynamics of the antibody.”
Mutations in the antibody residues cause only small changes overall, as they occur outside the protein domain, with non-covalent binding resulting in secondary disturbances.
In the N501Y variant, a greater number of changes in flexibility occur in the BD23-Fab complex as compared to the B38-Fab complex. The increase in rigidity was more pronounced for BD23. This is likely due to its smaller size that causes the N501Y mutation to experience an exaggerated dynamic motion propagated through the whole of the antibody molecule.
For the beta variant, the co-occurrence of K417N and E484K counter this effect by bringing the RMSF down near to that of the wildtype. With the kappa strain, L452R and E484Q reduce antibody flexibility, especially in the distant domain.
Allosteric effects
The B38-Fab complex also revealed large changes in correlation to the antibody residues with mutations in the RBD and adjacent residues. Taken together, the mutation of a single RBD residue can perturb antibody-RBD binding strongly and over a long-range.
This finding remained true for the mutations of N501Y, N417 in the K417N-E484K-N501Y mutant, R452, and Q484 in the L452R-E484Q variant, as well as the neighbors of each mutant. This indicates an allosteric information flow between the antibody and RBD.
In BD23-Fab with the K417N-E484K-N501Y mutant, the same change was seen but to a smaller extent.
The change in correlation between antibody and RBD residues was pronounced in certain regions in the antigen-binding domain. In fact, specific residues exhibited significant changes as a result of both altered residues and those associated with the whole RBD.
While B38 binds more strongly to the alpha variant, BD23 shows the opposite trend, binding more strongly to the beta variant. Apparently, information flow between the binding and interacting residues is quite different between the two antibodies.
The role played by residues in allosteric information flow
Using a protein graph-connectivity network model, the researchers of the current study found that in B38-Fab, changes in allosteric information flow with these mutations mainly occurs in the antigen-binding domain residues.
The spread of the local correlation through the amino acid chains leads to dynamic changes over a long distance. More specifically, the residues 23-30 in chain B near N501Y appear to change to residues 92-98 in chain A near K417N and E484K/Q mutations.
The location of the mutation is key. The asymmetric nature of this switch in BD23-Fab leads to a change in how all antibody residues correlate with mutated RBD residues.
In the BD23-Fab, however, irrespective of the location of the mutation, a large part of the binding interface was found to lose centrality, especially over a region of the antigen-binding domain. This is seen with all mutants.
Here, though the allosteric signal is being transmitted long-range, the dynamic effect is generalized. That is, the change in the correlation of some antibody residues occurs with all RBD residues, as compared to only the mutated ones in B38.
”These contrasting features show that there can be inherent differences in the microscopic dynamics of the antibody, depending on the nature of mutations in the antigen and on the nature of the antibody. This can have repercussions beyond the specific SARS-CoV-2 spike antigen and to the fundamental process of antigen-antibody recognition in general.”
Dissociation
The 470-490 loop is key to the unbinding of the antibody-RBD complex. In fact, this loop is the last point of contact between the antibody and the virus, as it acts as an anchor to prevent complete loss of attachment to the antibody.
This loop also contains the residue E484. This is the last RBD residue to dissociate with the antibody as it loses its hydrogen bond. When the K/Q484 mutation is present, the E484 residue is replaced by the S477 and sometimes A475, in the same loop.
In B38-Fab, E484 and E484K/Q (Glu to Lys or Gln) mutations show similar interaction energy, as neither of these mutations are in direct contact with the antibody interface. Therefore, all mutations play a similar role in antibody dissociation.
Notably, the functions performed by these mutations are less effective than the wildtype RBD in detaching the virus from the antibody. This reduced activity is partly due to the N501Y mutation, which has fewer hydrogen bonds than the other mutations. The N501Y is therefore a destabilizing mutation for RBD-B38.
The opposite occurs with BD23-Fab, where E484 is at the interface of binding. Thus, it is key to stabilize the complex via hydrogen bonding. The K/Q mutations here reduce its ability to form hydrogen bonds steeply, thereby resulting in a sharp reduction in the work required for antibody detachment.
Conclusion
“Future studies in this direction can lead to a deeper understanding of the immune evasion mechanism of mutating viruses and can help in designing adaptive therapeutic strategies against new viral strains by immunization through monoclonal antibodies and vaccination.”
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
 Computationally designed antibody attacks previously untreatable KRAS cancer mutations
Computationally designed antibody attacks previously untreatable KRAS cancer mutations