SARS-CoV-2 is a generalist virus that infects and evolves in multiple mammals, such as companion and captive animals, humans, and wild animals. Transmission across non-human species increases the chance of SARS-CoV-2 reservoirs forming, making eradication more challenging, and offers the virus with possibilities of novel evolutionary pathways, like the emergence of new mutant lineages and the selection of adaptive mutations.
Numerous studies demonstrated that SARS-CoV-2 transmits from humans to specific animal species and, in certain instances, backward to humans. However, despite the fact that these case reports have been helpful in identifying patterns of coronavirus disease 2019 (COVID-19) transmission and adaptability among particular animal species, a worldwide, systematic analysis of the present data is lacking.
About the study
In the current work, the researchers thoroughly investigated SARS-CoV-2 transmission among mammalian species, both human and non-human, to uncover mutations linked with each species using phylogenetic analyses and publicly accessible viral genome sequences.
Utilizing a large dataset, the team systematically examined SARS-CoV-2 sequences generated from animal hosts. They used the Global Initiative on Sharing All Influenza Data (GISAID) to retrieve all existing SARS-CoV-2 genomic sequences obtained from non-human animals.
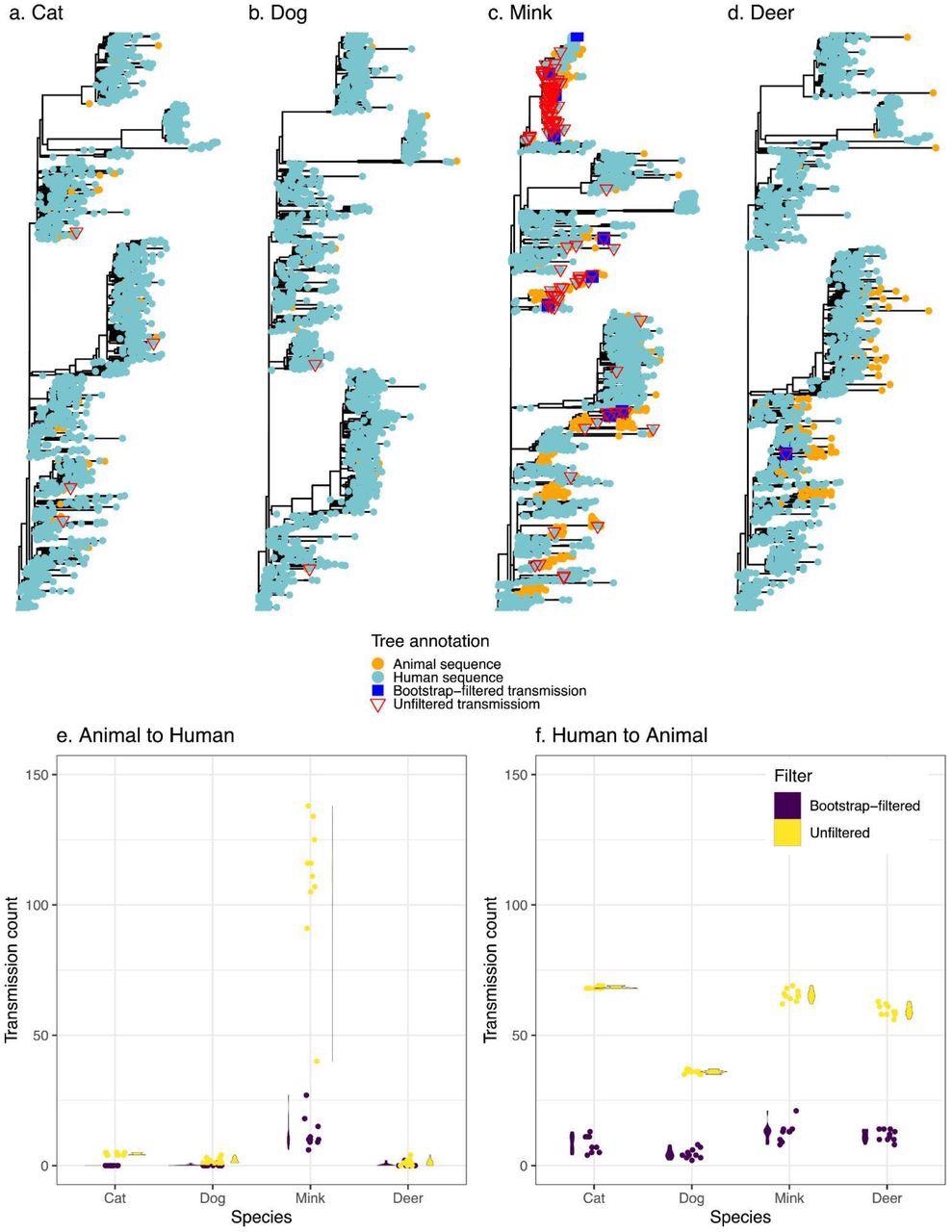
Transmission events inferred between humans and animals. Panels a-d display a representative tree for every species with animal to human transmissions marked on the tree. Trees are rooted with the Wuhan reference genome. Panels e and f display the distribution (violin plots alongside points plotted with jitter to avoid overlap) of inferred transmission counts (across 10 replicate trees) in each animal species, in both bootstrap-filtered and unfiltered trees.
The scientists estimated the relative frequencies of transmission among humans and four other often afflicted animals (Felis catus domesticus (cats), Canis lupus familaris (dogs), Odocoileus virginianus (deer), and minks) using phylogenetic approaches. They employed ancestral state restoration on viral phylogenetic trees to thoroughly explore the probable animal-to-human transmission incidents, including the transmission direction. The team's goal was to develop a uniform comparative methodology for interspecies transmission rather than extrapolate absolute frequencies of cross-species transmission of COVID-19.
The investigators used genome-wide association studies (GWAS) to investigate mutations linked to specific animal species relative to humans. They used POUTINE, a technique that implicitly regulates population structure and interconnection between mutations by addressing just homoplasic mutations that are similar by state and not by descent, which occurs independently numerous times within the phylogeny for conducting GWAS. Besides, the researchers performed a GWAS separately to discover variants correlated with each species.
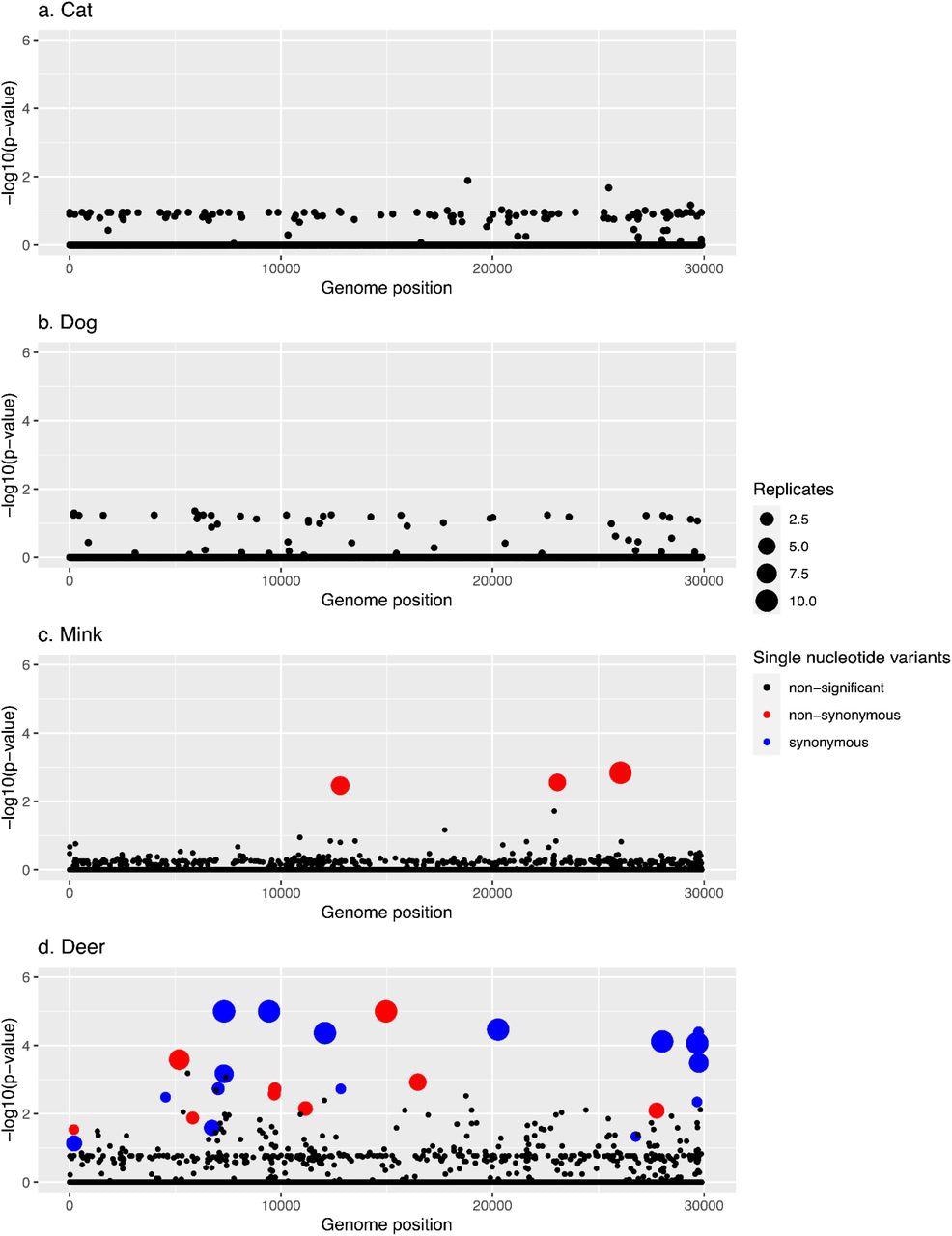
Manhattan plots summarizing GWAS hits in each animal species. In every panel, the x-axis represents the nucleotide position in the SARS-CoV-2 reference genome and the y-axis represents the -log10 of the pointwise p-values averaged over replicates. Statistically significant hits with family-wise corrected p-values of lower than 0.05 are shown in red (non-synonymous) or blue (synonymous), while non-statistically significant p-values are in black.
Results
The authors discovered that mink had the highest rate of animal-to-human SARS-CoV-2 transmission, agreeing with previous literature. On the contrary, the other examined species like dogs, cats, and deer had insignificant levels of SARS-CoV-2 transmission to humans. However, despite these findings, the possibility of SARS-CoV-2 transmission from cats or dogs to people cannot be ruled out, which could be more easily discovered with prospective household transmission research or deeper sampling.
The transmission of SARS-CoV-2 from humans to animals was consistent across species, and the slight discrepancies might be due to different sampling approaches. Although potential sampling biases may restrict predicted transmission events, the present findings serve as a valuable baseline for future research.
The team recovered the N501T mutation priorly linked with mink and two additional amino acid variations in other SARS-CoV-2 genes. They also discovered several unique deer mutations, including non-synonymous and synonymous substitutions. None of the single nucleotide variants (SNVs) were substantially linked with dogs or cats in GWAS, while 26 SNVs closely correlated with deer and three with mink.
Indeed, one of the three hits in minks appears in all 10 replicates, and at least 50% of the replicates harbored the remaining two. The three mutations, including N501T, were non-synonymous. Further, seven of the 26 hits in deer appeared in all 10 replicates, and five of them were present in at least 50% of the runs. Five of the 26 hits were intergenic mutations, whereas 12 were synonymous mutations. Additionally, 21 of the hits were cysteine (C)>uracil (U) transition mutations.
Conclusions
Together, the study findings uncovered a significant frequency of mink-to-human COVID-19 transfer incidents, while animal-to-human COVID-19 transmission from dogs, cats, and deer was uncommon. The team noted that future investigations should look into the SNVs strongly correlated with deer and minks to see if they harbor a function in variable pathogenesis, immune evasion, or modification of the host response.
The current findings support prolonged deer-to-deer COVID-19 transmission and emphasize the necessity of researching animal-related SARS-CoV-2 mutations to determine their potential influence on animal and human health. From accessible genomic sequences, the present work offers a quantitative methodology for monitoring COVID-19 transmission between animals and identifies numerous possible animal-adaptive mutations for further investigation.
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
Zooanthroponotic transmission of SARS-CoV-2 and host-specific viral mutations revealed by genome-wide phylogenetic analysis; Sana Naderi, Peter Chen, Carmen Lia Murall, Raphael Poujol, Susanne A Kraemer, Bradley S Pickering, Selena M Sagan, B. Jesse Shapiro. bioRxiv preprint 2022. DOI: https://doi.org/10.1101/2022.06.02.494559, https://www.biorxiv.org/content/10.1101/2022.06.02.494559v1
- Peer reviewed and published scientific report.
Naderi, Sana, Peter E Chen, Carmen Lia Murall, Raphael Poujol, Susanne Kraemer, Bradley S Pickering, Selena M Sagan, and B Jesse Shapiro. 2023. “Zooanthroponotic Transmission of SARS-CoV-2 and Host-Specific Viral Mutations Revealed by Genome-Wide Phylogenetic Analysis.” Edited by C Brandon Ogbunugafor, Detlef Weigel, and Nash Rochman. ELife 12 (April): e83685. https://doi.org/10.7554/eLife.83685. https://elifesciences.org/articles/83685.
 Genetic mapping in wastewater sets a new standard for pathogen surveillance
Genetic mapping in wastewater sets a new standard for pathogen surveillance