 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Background
Protease inhibitors are one of the most potent antiviral agents. The United States (US) Food and Drug Administration (FDA) recently approved the use of Paxlovid (ritonavir/nirmatrelvir), the first protease inhibitor against the SARS-CoV-2 main protease, such as 3CLpro and non-structural protein 5 (Nsp5).
Mouse hepatitis virus (MHV) 3CLpro was utilized as a stand-in for SARS-CoV-2 3CLpro to gather resistance information in the investigations that led to the Paxlovid Emergency Use Authorization (EUA). The 3CLpro of MHV and SARS-CoV-2 show a 50% sequence similarity. Notably, there is currently no published evidence on SARS-CoV-2 resistance.
Remarkably, novel SARS-CoV-2 variants outperformed the current coronavirus disease 2019 (COVID-19) vaccines. Hence, a similar threat exists for future COVID-19 antiviral medications.
About the study
The present study compared SARS-CoV-2 3CLpro and a panel of distinct viral proteases encompassing MHV 3CLpro in a recently established gain-of-signal experiment to establish Paxlovid-resistant SARS-CoV-2 3CLpro mutants.
The team created a chimeric Vesicular Stomatitis Virus (VSV) variant, replacing the intergenic domain between the polymerase (L) and glycoprotein (G) with the SARS-CoV-2 3CLpro. This was to establish a safe substitute for gain-of-function investigations.
For the initial proof-of-concept analysis, the researchers selected a mutant targeting the inhibitor GC376, harboring amino acid variation of phenylalanine to leucine at position 305 (F305L) in the 3CLpro. Further, the F305L mutant was employed in later mutation-selection research to analyze the possibility of several mutations accumulating in the 3CLpro.
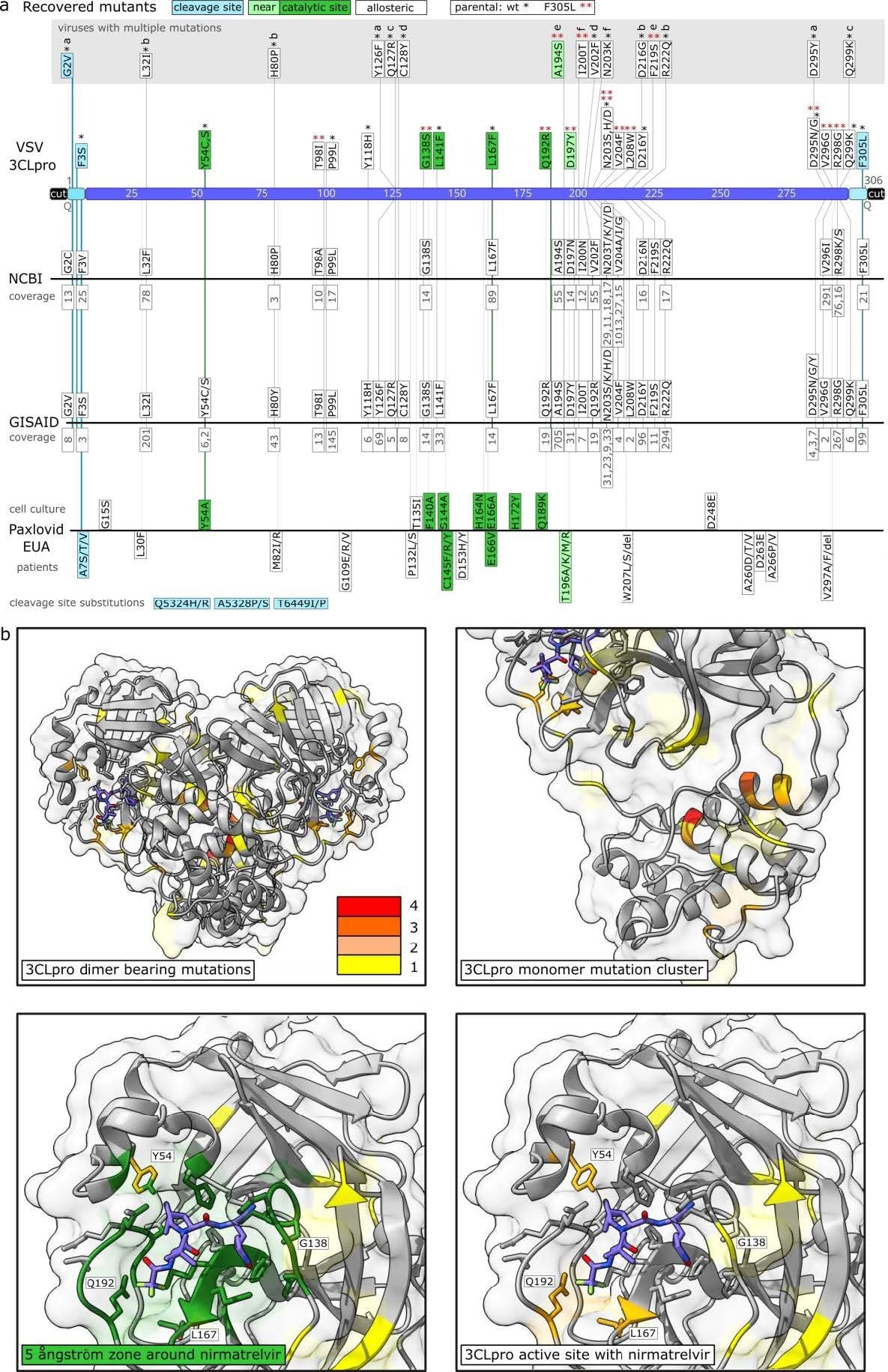
Sequencing of escape mutants and comparison to sequence data bases and Pfizer EUA. a: Mutants were recovered from VSV-G-3CLpro-L wild-type (*) and pre-mutated F305L variant (red **). Autocleavage site mutants are colored in turquoise, catalytic site mutants in green, near catalytic site mutants in light green and “allosteric” mutants in white. Viruses with more than one mutation are displayed separately and listed a-f. Frequencies from sequence data bases are displayed below the mutations in grey. Mutations from Pfizer EUA are divided into mutations found in cell culture and such sequenced from treated patients. Additional distant cleavage site mutations outside the 3CLpro open reading frame are depicted at the bottom. The coverage of mutation entries was obtained on march 3rd, 2022. b: Visualization of mutation-affected residues. Residues that were mutated one time are highlighted in yellow, two times in light orange, three times dark orange and four times in red. The 3CLpro protease dimer with bound nirmatrelvir was visualized in ChimeraX from the Protein Data Bank structure 7vh8. Catalytic center mutations are within a range of 5 ångström as visualized in dark green.
In addition, the authors determined nirmatrelvir-resistant 3CLpro using the SARS-CoV-2 F305L mutant and wild-type strains. Subsequently, they examined the 3CLpro and nearby G and L regions across the selected viruses.
The scientists classified the mutations found throughout the 3CLpro sequence into autocleavage and catalytic site mutants. Allosteric mutants were selected as the third category for any mutations that did not fall into the first two. They searched for those particular mutants in the Global Initiative on Sharing All Influenza Data (GISAID) EPICOV and the National Center for Biotechnology Information (NCBI) virus database.
GISAID depositions made before and following the EUA of Paxlovid on 22 December 2022 were separated into two groups. The researchers selected six virus samples to execute replication kinetics and dose-response studies relative to their ancestral virus variant to validate the putative resistance mutations revealed. They chose viruses for additional assessments centered on two criteria. Initially, the authors selected viral strains that have catalytic site alterations. Second, they included the most prevalent mutant outside the catalytic core.
The team reintroduced a few catalytic center mutations to a recently established protease function evaluation tool relying on replication-incompetent VSV. This was to substantiate the replication-competent VSV-G3CLpro-L's resistance findings while excluding the possibility of any new mutations developing throughout the dose-response trial. The read-out method was switched from a FluoroSpot to a fluorescence-activated single-cell sorting (FACS) to increase the test sensitivity.
Results and conclusions
The study results demonstrated that in the recently established gain-of-signal test, MHV 3CLpro responds to nirmatrelvir only modestly. Although the structures of the MHV and SARS-CoV-2 3CLpro were significantly conserved, the scientists hypothesized that the low amino acid sequence identity changes the binding pocket affinity towards nirmatrelvir considerably to produce some resistance against the inhibitor. Within 5 ngström or less, the binding pocket's key matching residues: M165-L, H164-Q, P168-S, R188-A, V186-R, A191-V, and T190-V, were different.
Additionally, the MHV 3CLpro sequence already has the amino acid alterations F305L and Y126F observed in the mutation induction experiment. Therefore, the researchers contend that MHV 3CLpro was not the best surrogate for 3CLpro resistance testing.
The mutant F305L was found in a small proof-of-concept analysis using the inhibitor GC376 and exhibited resistance to GC376 and nirmatrelvir and a slightly rapid replication kinetic. The adjustment of the C-terminal autocleavage zone from FQ (P2/P1) towards LQ (P2/P1), which was a favored recognition motif, may account for these features.
Mutations from the SARS-CoV-2 F305L and wild-type strains were detected using selection pressure for nirmatrelvir, enabling the mutants to bypass the inhibitor. The resistance phenotype of these mutations was confirmed by conducting dose-response tests and introducing mutations again into recently established protease activity monitoring systems.
Mutants respond differently to the inhibitor compounds because of their structural difference, as the team demonstrates using the examples of nirmatrelvir and GC376. The present findings might assist the directed compound structure adaptation in the future.
Collectively, the SARS-CoV-2 3CLpro mutations responsible for the resistance to the protease inhibitor nirmatrelvir, such as G138S, Y54C, L167F, A194S, Q192R, and F305L, were discovered in the current investigation. These results necessitate the cautious use of protease inhibitors in high-risk patients, as extensive non-selective usage could result in drug resistance rapidly.
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
SARS-CoV-2 3CLpro mutations confer resistance to Paxlovid (nirmatrelvir/ritonavir) in a VSV-based, non-gain-of-function system; Emmanuel Heilmann, Francesco Costacurta, Andre Volland, Dorothee von Laer. bioRxiv preprint 2022, DOI: https://doi.org/10.1101/2022.07.02.495455, https://www.biorxiv.org/content/10.1101/2022.07.02.495455v1
- Peer reviewed and published scientific report.
Heilmann, Emmanuel, Francesco Costacurta, Seyed Arad Moghadasi, Chengjin Ye, Matteo Pavan, Davide Bassani, Andre Volland, et al. 2023. “SARS-CoV-2 3CL pro Mutations Selected in a VSV-Based System Confer Resistance to Nirmatrelvir, Ensitrelvir, and GC376.” Science Translational Medicine 15 (678). https://doi.org/10.1126/scitranslmed.abq7360. https://www.science.org/doi/10.1126/scitranslmed.abq7360.
 Scientists identify common symptom patterns in post-COVID-19 vaccination syndrome
Scientists identify common symptom patterns in post-COVID-19 vaccination syndrome