 *Important notice: bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
*Important notice: bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
Background
Analysis of mutations around the LA binding RBD pocket in the SARS-CoV-2 Omicron variant S protein suggests that alterations may occur in locked conformation accessibility without LA binding at a neutral pH. Further, ionizable group mutations in the Omicron S protein could further alter pH dependence beyond that which is associated with the D614G mutation.
The SARS-CoV-2 S trimer can exist with its monomers closely/tightly packed, which is otherwise referred to as locked conformation. The locked S conformation is associated with LA binding at neutral pH. At an acidic pH, a similar S conformation may exist without LA binding.
Although several studies have deciphered S trimer structurally, mechanisms working behind its conformation's pH-dependence and how that relates to its function remain relatively unknown. Furthermore, there remain a lack of information available on how mutations in the S protein change the relative stability of its locked, closed (RBD down), and open (RBD up) conformations.
About the study
The present study examined a range of S RBD structures of coronaviruses, including those with D614G mutations such as the SARS-CoV-2 Delta and the Omicron variants. The researchers also closely inspected the LA binding RBD pocket to observe the open (RBD up) and closed (RBD down) conformations.
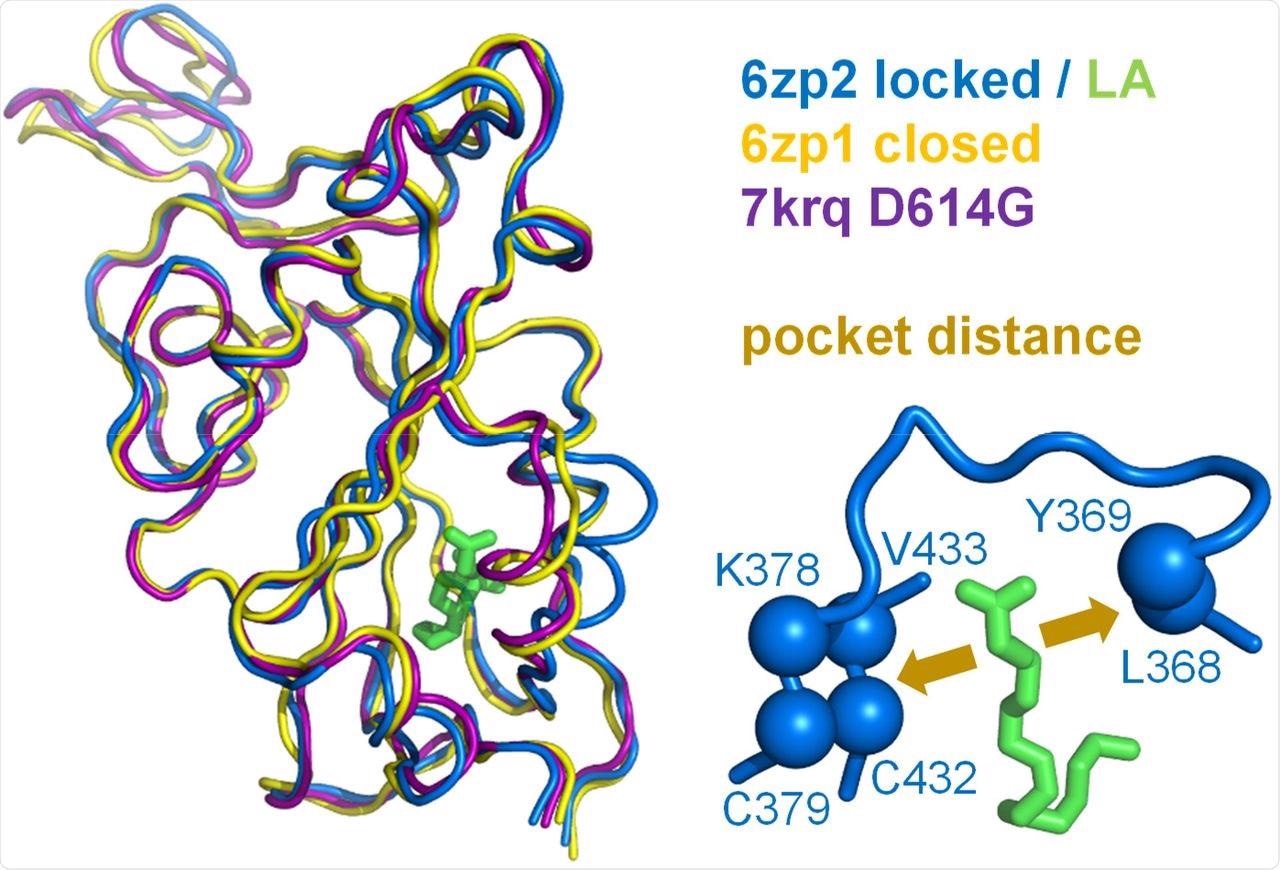
A measure of pocket opening in the RBD. Receptor binding domains are aligned for representative locked (6zp2) and closed (6zp1) (Xiong et al., 2020) S protein trimers, and also for an S trimer structure carrying the D614G mutant, 7krq (Zhang et al., 2021a), with all RBDs down and monomer burial approaching the locked form. Distance across the pocket is shown schematically for 6zp2, with linoleic acid (LA) bound. The distance is calculated between the average of the 4 β-strand Cαatoms displayed (left of LA, which align well structurally between these RBDs), and the average of the two Cα atoms shown to the right of LA, present on a turn within a sub-structure that gates the LA binding pocket (Toelzer et al., 2020).
To this end, they observed that these conformations differ by the distance between the average of the four β-strand Cα atoms on one side of the RBD pocket (amino acids C379 and C432) and the average of the two Cα atoms towards the opposite side (amino acids L368, Y369). The possibility of the D614G mutation around the RBD of the Omicron S protein leading to LA binding pocket changes and alter pH dependence was also examined.
Study findings
Several previous studies have calculated the changes in solvent accessible surface area (d-SASA) upon S monomer incorporation in various trimer forms. There is data supplementing two recent structures of the Delta variant identifiers 7v7n and 7sbk, as well as sequence changes around the RBD pocket of the Omicron S protein.
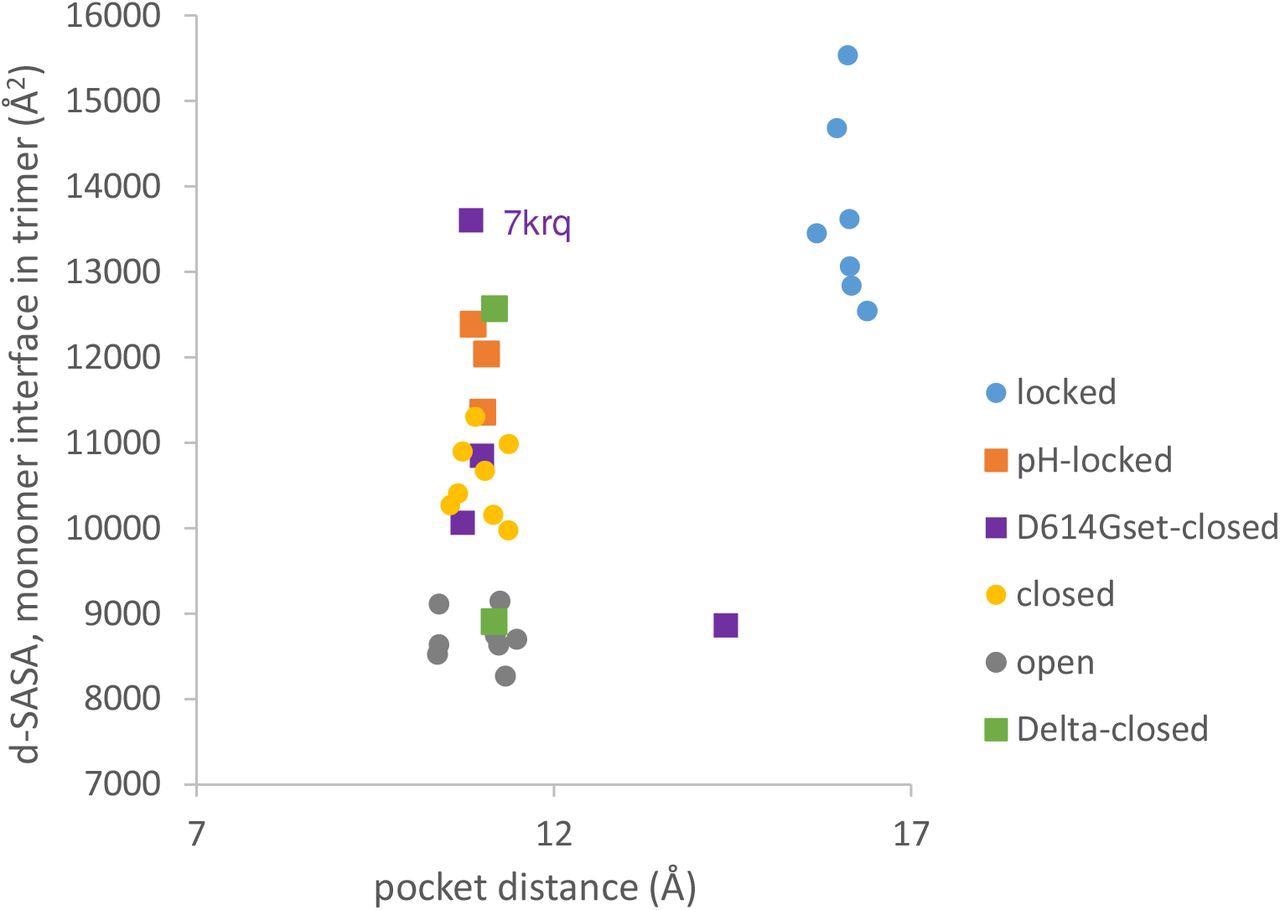
Variants carrying the D614G mutation do not cluster on a plot of pocket distance against S monomer burial within the S trimer. Clustering of locked, pH-locked, closed, and open forms in terms of monomer burial (Lobo and Warwicker, 2021) extends also to pocket distance. However, D614G set S proteins show greater variation and the two Delta variant S proteins displayed are also well separated in monomer burial. Values are 3 monomer averages for each of the S protein trimers. Other than the open set, only trimers with all RBDs down are included (and thus sets are named D614Gset-closed and Delta-closed).
The present study results revealed that locked forms are distant with a larger occupied RBD pocket and high monomer burial. The scatter plots comparing the RBD pocket distance and monomer interface showed that open and closed conformations cluster with a distance consistent with a closed RBD pocket at low (open) and intermediate (closed) monomer burial in the trimer. Importantly, the D614G substitution reduces the barrier between locked and closed S conformations at neutral pH.
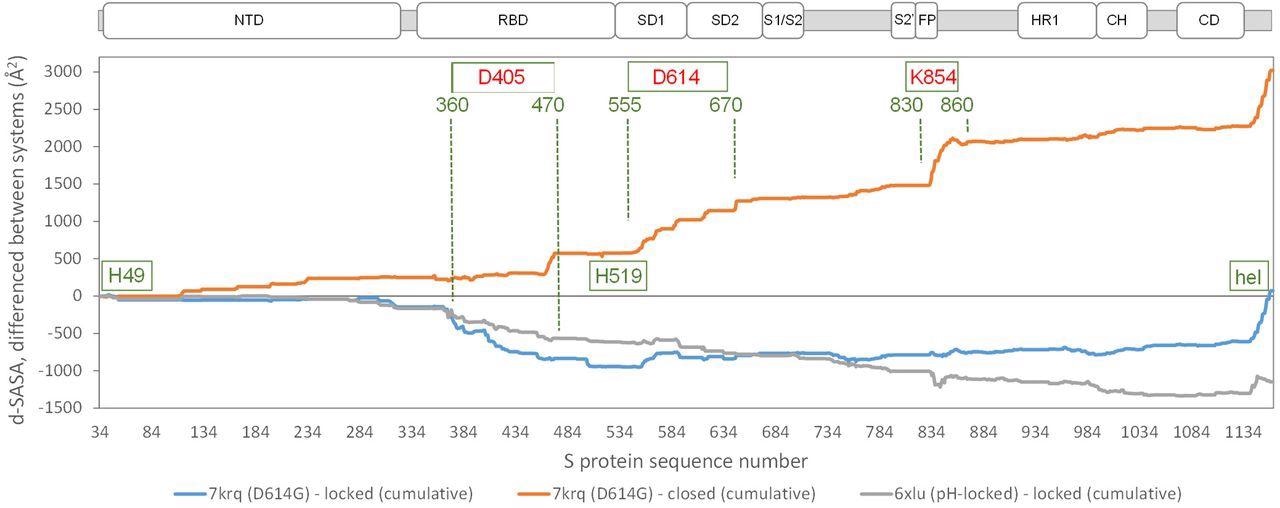
Comparison of acidic pH and D614G mutation effects on packing within the S trimer. The d-SASA value (monomer burial within a trimer) is further differenced between the systems indicated, between 7krq (D614G, closed pocket but burial approaching the locked form) and the average over the locked set, between 7krq and the average over the closed set, and between 6xlu (pH-locked) and the average over the locked set. These double-difference quantities are plotted cumulatively over the sequence of the S trimer, for which structure is available, with sub-domains (Berger and Schaffitzel, 2020) indicated (NTD N-terminal domain, SD1/SD2 subdomains 1 and 2, S1/S2 proteolytic cleavage site between subunits, S2’ cleavage site within S2 subunit, FP fusion peptide, HR1 heptad repeat 1, CH central helix, CD connector domain). Regions and amino acids of particular interest are displayed with residue numbers and in relation to the changes in burial. A C-terminal helical coil interaction between monomers (present only in some structures) is labeled (hel).
In the SARS-CoV-2 variants carrying the D614G mutation, a salt bridge is lost between D614 and K854. This results in the formation of open (RBD up) forms, which enhances interaction with the receptor. The D614 mutation also appears to stabilize the S protein trimer against dissociation.
Another amino acid region between 360-470 residues showed the most differentiation between the D614G 7krq structure and the locked/pocket occupied form. This structure houses the LA pocket, including the gating mechanism and carboxylate groups of D405, D420, and E465 that also contribute to couple burial and pH-dependent stability together with D614.
Further, it was observed that the D614G S trimer mutation in the Omicron variant leads to RBD alterations and that the degree of RBD burial can vary substantially with fewer changes in the RBD pocket distance. Moreover, other non-ionizable group mutations such as S371L, S373P, and S375F in the sequence next to the RBD pocket gate mediate the escape of an immune response primed by vaccine or prior infection.
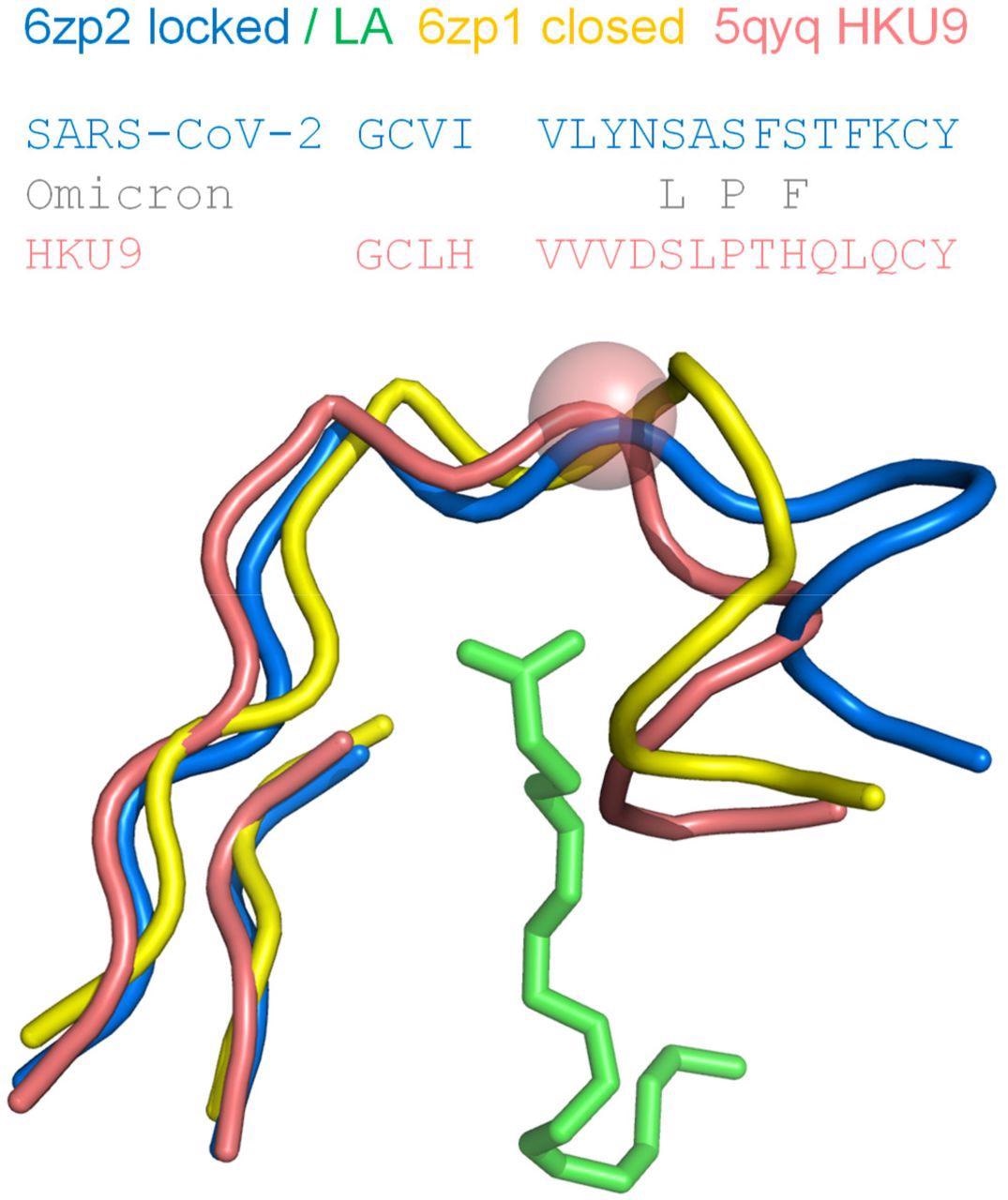
Proline in the RBD of coronavirus HKU9, a site equivalent to S373 in the SARS-CoV-2 RBD, exhibits a kink in the mainchain. Regions around the LA binding site of SARS-CoV-2 S protein RBD are shown for locked (6zp2) and closed (6zp1) structures, with LA from 6zp2. The β-strand elements align well structurally, whereas the difference in pocket gating structures between pocket open/occupied (6zp2) and closed/unoccupied (6zp1) is clear in the turn region at the right-hand side. A proline in the RBD of bat-derived coronavirus HKU9 (Huang et al., 2016), at the equivalent location to S373 of SARS-CoV-2 S protein, is indicated with a sphere at the Cα added to its tube mainchain. Amino acid sequences are shown for the contiguous turn-strand, with changes in the SARS-CoV-2 Omicron variant added.
It was also observed that a proline (S373P) residue lying in a segment connecting two sides of the LA pocket in the RBD offered reduced mainchain conformational freedom to the Omicron S trimer, which resulted in a bias towards a closed pocket conformation. Thus, changes in this region are likely to impact both the RBD pocket and the predictions of pH dependence.
Based on dSASA calculations, the RBD alterations of the S trimer were interpreted in two ways. As per the aspartimide/glutarimide (Asp/Glu) burial model, it would mean that either Y505H or D405N enriches neutral pH instability, thereby leading to a more closely packed RBD than that which has been observed for 7krq mutations of ionizable groups.
The second interpretation of these calculations is that some other mutations lead to lesser pH-sensing and remove tighter packing in this region. One common aspect for both interpretations on the changes in the Omicron S protein relative to D614G S protein is the maintenance of closer packing in the RBD C-terminal regions that is facilitated by the D614G mutation.
Conclusions
The current study analyzed mutations around the LA binding RBD pocket in the Omicron S protein. These findings suggest that further changes may occur in locked conformation accessibility at neutral pH, without LA binding. Furthermore, ionizable group changes could contribute to altered pH-dependence beyond that which is associated with D614G.
The current study also tested several hypotheses, such as the role of Asp/Glu sidechains on observed pH-dependent effects and histidines H49 and H519 mediating pH dependence.
More importantly, the authors of the current study suggest that it is not possible to use the RBD pocket distance as a guide to choosing one interpretation over the other. This is because the degree of RBD burial in the S trimer can vary substantially with little change in the pocket distance. Furthermore, there are other non-ionizable group mutations in this region of the Omicron S protein that should be considered.
*Important notice: bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
 Genetic mapping in wastewater sets a new standard for pathogen surveillance
Genetic mapping in wastewater sets a new standard for pathogen surveillance