The coronavirus disease 2019 (COVID-19), which is caused by infection with the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has caused over 5.44 million deaths as of January 4, 2022.
Although SARS-CoV-2 primarily impacts the respiratory system, recent research indicates that COVID-19 is also associated with neurological problems, with more than 30% of hospitalized COVID-19 patients displaying neurological symptoms. Magnetic resonance imaging (MRI) has confirmed the involvement of SARS-CoV-2 in the central nervous system (CNS) in both survivors and non-survivors in accordance with this observation.
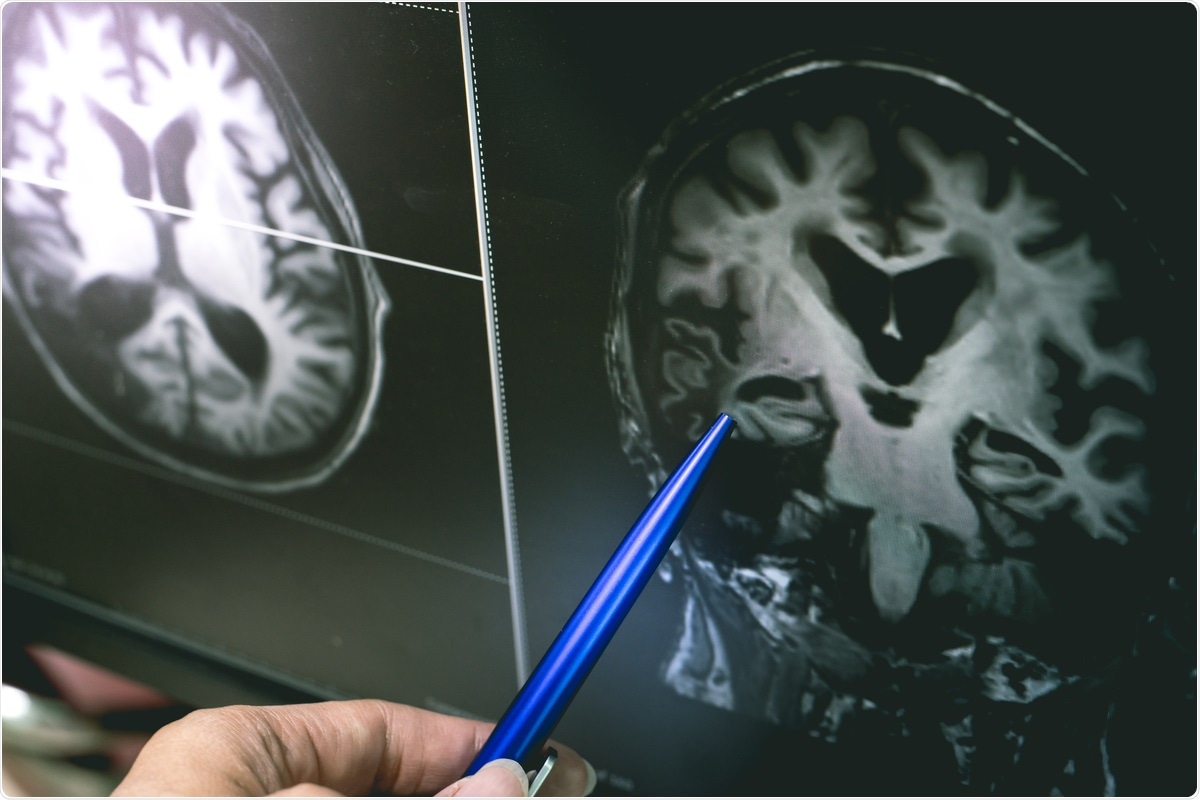
Study: Can SARS-CoV-2 Infection Exacerbate Alzheimer’s Disease? An Overview of Shared Risk Factors and Pathogenetic Mechanisms. Image Credit: Atthapon Raksthaput / Shutterstock.com
Background
Alzheimer's disease (AD) is the most prevalent type of dementia among the elderly globally. AD is clinically defined by neuronal death in the hippocampus and cortical areas, resulting in memory loss, behavioral abnormalities, and cognitive impairment.
Intracellular neurofibrillary tangles (NFTs), as well as parenchymal and vascular amyloid (Aβ) deposits, are all neuropathological hallmarks of AD. Neuroinflammation plays a critical role in the progression of the neuropathological alterations found in AD pathogenesis, which is attributed to activated microglia cells and the production of numerous cytokines.
Surprisingly, catastrophic consequences following SARS-CoV-2 infection in elderly adults are frequently associated with a cytokine storm that results in an overactive inflammatory and immunological response, which may accelerate brain inflammatory neurodegeneration.
In a recent Journal of Personalized Medicine review, researchers discuss the findings of previous studies on the role of SARS-CoV-2 infection in the progression of AD. AD and COVID-19 have a close association that could lead to the discovery of biomarkers for COVID-19 in people at high risk of acquiring AD, as well as assist in the management and development of novel therapeutics for both diseases.
Risk factors and pathogenic mechanisms shared by AD and COVID-19
Age
The greatest risk factor for AD is aging, with the disease's prevalence increasing every five years beyond the age of 65. In COVID-19 patients, aging is a well-known risk factor for severe disease and death.
In fact, the case fatality ratio (CFR) of COVID-19 increases with age, according to epidemiological data from China. More specifically, patients aged 59 years or older were at least five times more likely to die following the onset of symptoms than those younger than 59.
In Italy, which was the first country afflicted by the pandemic after China, the CFR of COVID-19 in patients over the age of 80 was almost two times greater than the overall population. Aging is thought to cause the creation of reactive oxygen species (ROS), worsen Aβ production, and enhance neuroinflammation, all of which contribute to the pathophysiology of COVID-19 and AD.
Aβ
Aβ is thought to be a critical mediator of AD pathogenesis and is one of the first brain AD-related molecular changes that can be detected several years before clinical symptoms appear. Some evidence suggests that Aβ may be linked to an increased risk of developing AD in COVID-19 patients.
The authors found that Aβ has better antibacterial efficacy against the most prevalent and clinically important pathogens. As a result, it is possible that SARS-CoV-2 infection stimulates or accelerates Aβ build-up in the brain as part of an innate immune response, leading to AD. Another study found that the SARS-CoV-2 spike S1 protein's receptor-binding domain (RBD) may bind to numerous proteins, including Aβ and tau, thus increasing their aggregation and continuing the neurodegenerative process.
ACE1 and ACE2
Angiotensin-converting enzymes (ACE), which include ACE1 and ACE2, are essential components of the renin-angiotensin system (RAS). These enzymes work in opposition to each other to regulate the levels of angiotensin II (Ang II) and angiotensin (1–7).
ACE1 converts Ang I to Ang II and inactivates the vasodilator peptide bradykinin. Comparatively, ACE2 is responsible for the cleavage of Ang II into smaller proteins such as Ang(1–7), which has antiproliferative and vasodilator properties.
Notably, several ACE inhibitors have been shown to prevent neurodegenerative illnesses, including AD, by decreasing Ang II levels through anti-inflammatory and antioxidant actions.
In contrast to ACE1 and Ang II, Ang(1–7) proteins bind to Mas receptor (MASR), thereby producing the ACE2/Ang(1–7)Mas axis, which is known to play a protective function in neurodegeneration. Ang(1–7) inhibits inflammatory and oxidative stress events by activating MASR, which regulates the activation of the PI3K/Akt/CREB/BDNF/TrKB pathway. The brain-derived neurotrophic factor (BDNF) is widely known for its function in neurogenesis and neurodevelopment, as well as mood regulation.
Conclusions
Despite the high risk of severe COVID-19 among the elderly, there are only a few studies that have been published addressing the relationship between COVID-19 and AD. However, the numerous shared links between these two diseases highlight the importance of assessing neurological symptoms and implementing preventive strategies to reduce the risk of developing AD in SARS-CoV-2 infected people.
To assess the long-term neurological effects of SARS-CoV-2 infection, longitudinal follow-up studies of COVID-19 patients are required. Furthermore, large-scale retrospective analyses combined with preclinical investigations will be necessary to completely comprehend the implications of SARS-CoV-2 infection on the onset and progression of AD.
Finally, in order to examine the pathogenetic pathways shared by AD and COVID-19, these investigations should include cognitive impairment evaluation, blood and neuroimaging biomarkers measuring inflammation, oxidative damage, or metabolic changes.
Journal reference:
- Villa, C., Rivellini, E., Lavitrano, M., & Combi, R. (2021). Can SARS-CoV-2 Infection Exacerbate Alzheimer’s Disease? An Overview of Shared Risk Factors and Pathogenetic Mechanisms. Journal of Personalized Medicine 12(1). doi:10.3390/jpm12010029.
 Scientists find that human embryos are vulnerable to COVID
Scientists find that human embryos are vulnerable to COVID