Stick, tag, destroy: The science of PROTACs and MGDs
Traditional drug research has focused on creating inhibitors that occupy the active areas of disease-related proteins, preventing them from functioning properly.
But this strategy leaves a considerable amount of the human proteome, estimated to be around 80 % of disease-related proteins, "undruggable", due to the absence of specific active sites or binding pockets.1,2
Targeted Protein destruction (TPD) may be a revolutionary alternative, using small chemicals to trigger ubiquitination and then destruction of target proteins via the cell's natural proteasomal or lysosomal pathways.2
Within the TPD landscape, Proteolysis-Targeting Chimeras (PROTACs) and Molecular Glue Degraders (MGDs) stand out as the leading strategies1-3.
PROTACs (Proteolysis-targeting chimeras)
A PROTAC molecule is typically a heterobifunctional compound made of three important parts: a ligand that binds to the protein of interest (POI), another ligand that recruits an E3 ubiquitin ligase, and a chemical linker that joins the two (Figure 1).4,5
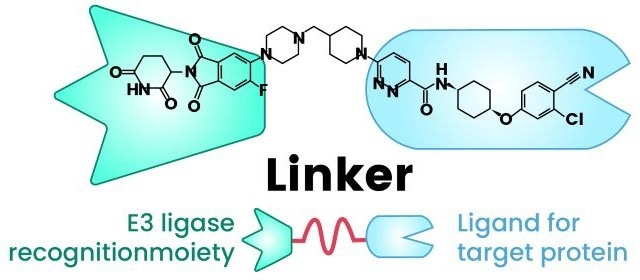
Figure 1. Structure of a PROTAC. The molecule links an E3 ligase-binding domain to a target protein ligand via a flexible chemical linker, enabling targeted ubiquitination and subsequent proteasomal degradation6. Image Credit: PROTAC, A "Revolutionary" Technology in Small Molecule Drug Discovery- CUSABIO
PROTAC activity is based on its capacity to bring the POI and an E3 ligase into close proximity with each other, resulting in a ternary complex. This closeness promotes the transfer of ubiquitin tags from the E2 conjugating enzyme (which interacts with the E3 ligase) to the POI.2,3
Once polyubiquitinated, the 26S proteasome identifies and quickly destroys the POI (Figure 2). A significant advantage is that after the target protein is destroyed, the PROTAC molecule is released and regenerated to target another copy of the POI, allowing for continued degradation even at catalytic drug concentrations.1,2
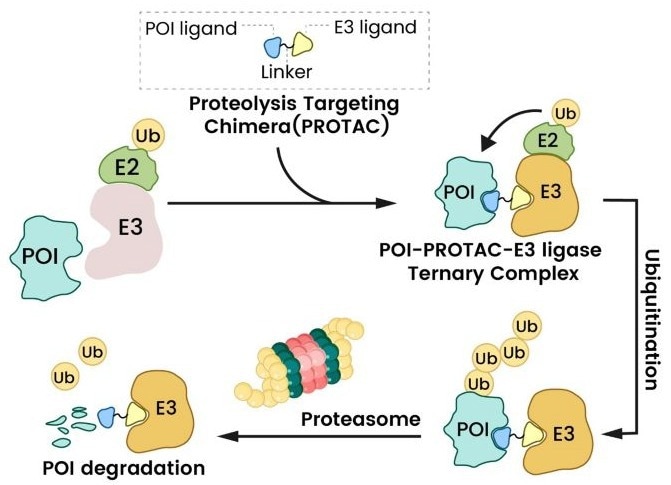
Figure 2. PROTAC mechanism of action. The PROTAC bridges a target protein and E3 ligase, triggering ubiquitination and proteasomal degradation1. Image Credit: Sino Biological Inc.
Historical development and clinical progress
The concept of PROTACs was established in 1999, with the first peptide-based PROTAC molecule (PROTAC-1) created in 2001.7 These early PROTACs demonstrated the notion of targeted protein degradation, but were restricted by low cell permeability, limited oral bioavailability, and proteolytic instability.
The first entirely small molecule PROTAC, PROTAC 4, was introduced in 2008. It demonstrated significantly improved cell permeability and paved the way for finding small-molecule ligands for important E3 ligases.8
Cereblon (CRBN) sprang to prominence in 2010 as the principal target of thalidomide and its analogs. Von-Hippel-Lindau (VHL)-recruiting ligands were later found in 2012.1-3 CRBN and VHL are now the most commonly used E3 ligases in PROTAC design due to their drug-like characteristics.
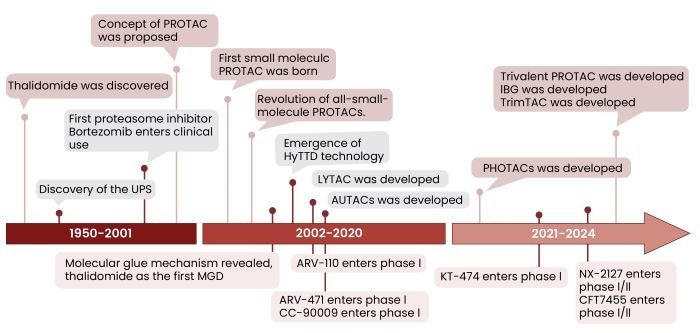
Figure 3. Key milestones in PROTAC development, from its initial conceptualization and peptide-based molecules to the first small-molecule PROTACs and the emergence of CRBN and VHL as E3 ligase targets2. Image Credit: Sino Biological Inc.
Since then, there has been rapid clinical development in the area, with numerous PROTACs entering clinical trials. Bavdegalutamide (ARV-110), an androgen receptor (AR)-targeting PROTAC, has completed Phase II studies for prostate cancer.9
Vepdegestrant (ARV-471), which targets the estrogen receptor (ER), has advanced to NDA/BLA for ER+/HER2− breast cancer.2,10 NX-2127, a Bruton's tyrosine kinase (BTK) degrader, has demonstrated efficacy in overcoming treatment resistance in B-cell malignancies.11
There are over 30 heterobifunctional protein degraders currently studied in Phase I-III trials1. Table 1 lists the representative clinical-stage PROTACs, their target proteins, the E3 ligases they recruit, and their current clinical stages.
Table 1. Representative clinical-stage PROTACs, their target proteins, and the E3 ligases they recruit1-3. Source: Sino Biological Inc.
| Molecule |
Target Protein |
E3 Ligase |
Clinical Phase |
| ARV-471 |
ER |
CRBN |
NDA/BLA |
| ARV-766 |
AR |
CRBN |
Phase II |
| ARV-110 |
AR |
CRBN |
Phase II |
| DT-2216 |
Bcl-XL |
VHL |
Phase I/II |
| NX-2127 |
BTK |
CRBN |
Phase I |
| NX-5948 |
BTK |
CRBN |
Phase I |
| CFT1946 |
BRAF V600 |
CRBN |
Phase I |
| KT-474 |
IRAK4 |
CRBN |
Phase II |
Challenges and optimization
Despite their potential, PROTACs have a few issues that prevent their widespread adoption, such as complex structures and large molecular weight (typically >700 Da), which frequently place them outside of the "rule of five". This can cause decreased cell permeability, metabolic instability, and adverse pharmacokinetic (PK) profiles.2,12
Rational design approaches address these issues by focusing on linker and E3 ligase ligand optimization.13 Incorporating rigid structures, such as spirocycles or piperidines, into the linker can significantly improve degradation potency and oral bioavailability.2
Optimizing E3 ligands entails creating novel scaffolds, such as phenyl glutarimide (PG) derivatives and TX-16, which have higher stability and affinity than standard thalidomide derivatives.2,4,12,13
Molecular glue degraders (MGDs)
MGDs are small, monovalent molecules, as opposed to the bivalency of PROTACs. They work by initiating or maintaining new protein-protein interactions (PPIs) between an E3 ligase and a target protein (POI). This interaction marks the target protein for destruction using the ubiquitin-proteasome pathway.2,3
MGDs achieve this by changing the surface properties of substrate receptors and E3 ligases, resulting in "non-native" interactions that enhance ubiquitination and degradation.3,14
MGDs were discovered mostly through serendipitous observation or repurposing existing pharmacological compounds, making rational design difficult due to the unpredictable nature of these generated interactions.15
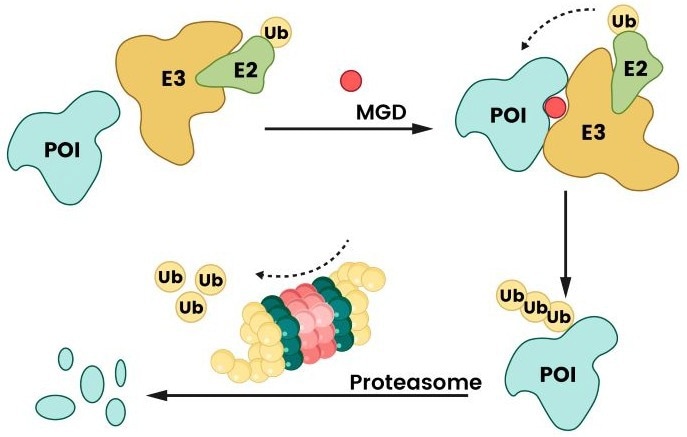
Figure 4. Degradation mechanism of MGD. MGD facilitates the interaction between a protein of interest (POI) and an E3 ligase by binding to either the E3 ligase or the POI, thereby promoting ubiquitination and subsequent proteasomal degradation2. Image Credit: Sino Biological Inc.
Advantages and challenges
MGDs have many encouraging characteristics in therapeutics. Their monovalent structure produces smaller molecules with lower molecular weights that are more easily subject to "Lipinski's rule of five". This often results in greater drug-likeness and pharmacokinetic characteristics than PROTACs.2
In addition, MGDs may not always require a binding pocket on the target protein, which broadens the list of "undruggable" proteins they can degrade.1,2
Thalidomide, lenalidomide, and pomalidomide were among the first MGDs identified and are now licensed to treat erythema nodosum, myelodysplastic syndromes, and multiple myeloma, respectively.1,2
Other MGDs, including CC-90009 (targeting GSPT1) and E7820 (targeting RBM39), are also in clinical trials.2,3
Designing sensible MGDs is still a challenge in this area. Their mechanisms are fundamentally unpredictable, and clinical translation faces challenges such as varying degradation efficiencies across tissues, a lack of prognostic biomarkers, and unexpected off-target consequences.
Recent efforts in rational MGD design have included tactics such as adding covalent handles to existing inhibitors, which effectively convert them into degradation-competent MGDs.1-3,12,15
Conclusion and future outlook
TPD technologies, particularly PROTACs and MGDs, represent a paradigm shift in drug development by completely eliminating target proteins rather than merely inhibiting them.
This ability to destroy "undruggable" proteins opens up new therapeutic paths for various disorders, particularly cancer.
Despite impressive progress, problems remain, such as enhancing metabolic stability, addressing potential off-target effects, and broadening the range of accessible E3 ligases beyond the currently dominating CRBN and VHL.
The combination of modern computational tools, such as AI-aided drug design, with comprehensive analytical methodologies, such as multi-omics profiling, is expected to speed the discovery of novel degraders while improving selectivity and efficacy.2,3
PROTAC and MGD technologies promise to open new therapy options for diseases previously considered unattainable.
SignalChem Biotech’s contribution to targeted protein degradation research
As targeted protein degradation technologies evolve, SignalChem Biotech provides researchers with a comprehensive suite of recombinant E3 ligases, including essential molecules such as CRBN and VHL, and a diverse array of target proteins, antibodies, and functional assay reagents.
These high-quality, validated proteins and reagents allow for rapid screening, mechanistic probing, and optimization of novel PROTACs and molecular glue degraders.
Partial list of ubiquitin enzymes - E3
Source: Sino Biological Inc.
| Name |
Cat# |
Species |
Tag |
Expression |
Sequence |
| BIRC3, Active |
B280-380G |
Human |
GST |
Sf9 Cells |
Full Length |
| BIRC7, Active |
B281-380G |
Human |
GST |
Sf9 Cells |
Full Length |
| HERC4, Active |
H265-381G |
Human |
GST |
Sf9 Cells |
642-end |
| MGRN1, Active |
M287-380G |
Human |
GST |
Sf9 Cells |
2-end |
| RanBP2, Active |
R230-381H |
Human |
His |
E.coli |
2553-2838 |
| RNF34 (CARP), Active |
R296-380G |
Human |
GST |
Sf9 Cells |
Full Length |
| RNF34L (CARP2), Active |
R297-380G |
Human |
GST |
Sf9 Cells |
Full Length |
| TRIM37, Active |
T292-380G |
Human |
GST |
Sf9 Cells |
Full Length |
| WWP2, Active |
W297-380G |
Human |
GST |
Sf9 Cells |
Full Length |
| CBL Protein |
C272-381G |
Human |
GST |
E.coli |
1-375 |
References
- Pan, M., et al. (2025). Proteolysis‐Targeting Chimera (PROTAC): A Revolutionary Tool for Chemical Biology Research. Small Methods. https://doi.org/10.1002/smtd.202500402.
- Pan, Y., Wang, Y. and Gou, S. (2025). Proteolysis targeting chimera, molecular glue degrader and hydrophobic tag tethering degrader for targeted protein degradation: Mechanisms, strategies and application. Bioorganic Chemistry, (online) 161, p.108491. https://doi.org/10.1016/j.bioorg.2025.108491.
- Zhang, B., et al. (2025). Rational Design of Dual Degraders by Incorporating Molecular Glue Structural Features into PROTAC Degraders. Journal of Medicinal Chemistry. https://doi.org/10.1021/acs.jmedchem.5c00443.
- Ito, T. (2023). Protein degraders -from thalidomide to new PROTACs. Journal of Biochemistry/The journal of biochemistry. https://doi.org/10.1093/jb/mvad113.
- Rutherford, K.A. and McManus, K.J. (2024). PROTACs: Current and Future Potential as a Precision Medicine Strategy to Combat Cancer. Molecular cancer therapeutics, (online) 23(4), pp.454–463. https://doi.org/10.1158/1535-7163.mct-23-0747.
- Pagan, J., et al. (2013). Role of the Ubiquitin Proteasome System in the Heart. Circulation Research, 112(7), pp.1046–1058. https://doi.org/10.1161/circresaha.112.300521.
- Sakamoto, K.M., et al. (2001). Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proceedings of the National Academy of Sciences, 98(15), pp.8554–8559. https://doi.org/10.1073/pnas.141230798.
- Schneekloth, A.R., et al. (2008). Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorganic & Medicinal Chemistry Letters, (online) 18(22), pp.5904–5908. https://doi.org/10.1016/j.bmcl.2008.07.114.
- Chirnomas, D., Hornberger, K.R. and Crews, C.M. (2023). Protein degraders enter the clinic — a new approach to cancer therapy. Nature Reviews Clinical Oncology. https://doi.org/10.1038/s41571-023-00736-3.
- Hamilton, E.P., et al. (2022). ARV-471, an estrogen receptor (ER) PROTACdegrader, combined with palbociclib in advanced ER+/human epidermal growth factor receptor 2–negative (HER2-) breast cancer: Phase 1b cohort (part C) of a phase 1/2 study. Journal of Clinical Oncology, 40(16_suppl), pp.TPS1120–TPS1120. https://doi.org/10.1200/jco.2022.40.16_suppl.tps1120.
- Robbins, D.W., et al. (2024). Discovery and Preclinical Pharmacology of NX-2127, an Orally Bioavailable Degrader of Bruton’s Tyrosine Kinase with Immunomodulatory Activity for the Treatment of Patients with B Cell Malignancies. Journal of medicinal chemistry, 67(4). https://doi.org/10.1021/acs.jmedchem.3c01007.
- Pu, C., et al. (2023). Current strategies for improving limitations of proteolysis targeting chimeras. Chinese Chemical Letters, 34(6), pp.107927–107927. https://doi.org/10.1016/j.cclet.2022.107927.
- Zagidullin, A., et al. (2020). Novel approaches for the rational design of PROTAC linkers. Exploration of Targeted Anti-tumor Therapy, 1(5), pp.381–390. https://doi.org/10.37349/etat.2020.00023.
- Malone, M.L., et al. (2025). Application of DELs for E3 Ligase Ligand Discovery and Targeted Protein Degradation. DNA-encoded Library Technology for Drug Discovery, pp.134–156. https://doi.org/10.1039/9781788016032-00134.
- Liu, Y., et al. (2025). Routes to molecular glue degrader discovery. Trends in Biochemical Sciences. (online) https://doi.org/10.1016/j.tibs.2024.12.006.
About Sino Biological Inc.
Sino Biological is an international reagent supplier and service provider. The company specializes in recombinant protein production and antibody development. All of Sino Biological's products are independently developed and produced, including recombinant proteins, antibodies and cDNA clones. Sino Biological is the researchers' one-stop technical services shop for the advanced technology platforms they need to make advancements. In addition, Sino Biological offer pharmaceutical companies and biotechnology firms pre-clinical production technology services for hundreds of monoclonal antibody drug candidates.
Sino Biological's core business
Sino Biological is committed to providing high-quality recombinant protein and antibody reagents and to being a one-stop technical services shop for life science researchers around the world. All of our products are independently developed and produced. In addition, we offer pharmaceutical companies and biotechnology firms pre-clinical production technology services for hundreds of monoclonal antibody drug candidates. Our product quality control indicators meet rigorous requirements for clinical use samples. It takes only a few weeks for us to produce 1 to 30 grams of purified monoclonal antibody from gene sequencing.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.