An international team of scientists has learnt how to determine the spatial structure of a protein obtained with an X-ray laser using the sulfur atoms it contains. This development is the next stage in the project of a group led by Vadim Cherezov to create an effective method of studying receptor proteins. A detailed description of the study has been published in the journal Science Advances.
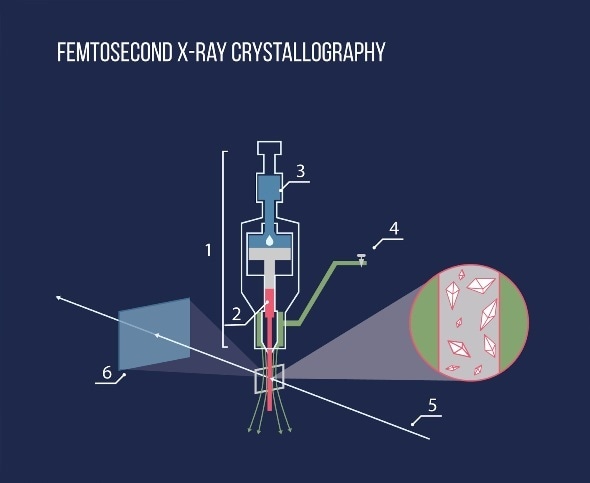
Diagram of the femtosecond X-ray crystallography experiment. Using the injector (1), the protein crystals dissolved in the lipid medium (2) are exposed to an X-ray beam (5), after which the scattered beam hits a detector (6). Pressure is created in the injector by a hydraulic piston (3), a special gas flow (4) is applied to maintain the straight shape of the lipid stream.
Receptor proteins (GPCRs) allow signals to be transmitted within cells, which, in turn, enables the cells to obtain information about their environment and interact with one another. As a result, we are able to see, feel, maintain blood pressure etc., i.e. everything that is needed for the functioning of our bodies. Any disorders in the way these proteins work can result in serious consequences, such as blindness. Developing medicines to restore the normal function of receptors is not possible without a precise understanding of the way in which GPCRs operate, which, as with other proteins, is determined by their spatial structure, i.e. the way in which the protein folds.
The best method of doing this is to use X-ray crystallography. For X-rays, a crystal is a three-dimensional diffraction lattice in which the radiation is scattered on the atoms.
A particular problem with this method is obtaining protein crystals. In order to do this, receptor proteins have to be extracted from a cell membrane and placed in a special lipid environment. Then, by selecting the temperature and using substances to speed up the nucleation process, the protein crystallizes.
One challenge with GPCRs is that they are highly mobile and dynamic molecules that frequently change their spatial structure. This means that it is difficult for them to grow large crystals that are needed for the classical diffraction procedure. This procedure involves exposing the crystal to radiation at different angles for a relatively long period of time. X-rays ionize the atoms, which destroys the protein molecules. Large crystals of a few dozen microns are what is needed to compensate for this effect.
Thanks to the new experimental method of Serial Femtosecond Crystallography, it is now possible to solve this problem. The method has been developed over the past few years by an international team of scientists from Arizona State University and the University of Zurich, SLAC National Accelerator Laboratory in Stanford, the iHuman Institute at ShanghaiTech University, the Institute of Biophysics of the Chinese Academy of Sciences, the CFEL center in Hamburg, the University of Southern California and MIPT. One of the leaders of the team is Vadim Cherezov, a professor at the University of Southern California and MIPT. The method is based on the use of new generation X-ray sources – free-electron lasers. The radiation they emit is so powerful that it fully ionizes atoms in a crystal as it passes through, essentially destroying it. However, as the laser pulse has a very short duration (a few femtoseconds, 10-15 s), a diffraction pattern can be recorded before the atoms move from their position. This has meant that the scientists are able to avoid the difficulties associated with radiation damage.
As the crystal is destroyed immediately, it is not possible to measure it at different orientations. To solve this problem, scientists collect and process data from several crystals. Using a special injector, the lipid environment in which the crystals are situated is exposed to an X-ray pulse. The whole process is similar to squeezing toothpaste out of a tube.
The result is millions of diffraction images that need to be processed: by selecting images with crystals, finding their orientation, and then putting them together in a three-dimensional diffraction pattern. Two parameters must be known in order to decipher the structure: the amplitude and the phase of the reflected radiation. The amplitude value is measured on a detector during the experiment, however determining the phase is a complex task and there are a number of ways of solving the problem.
For example, if we know of a certain protein that has a similar structure, we can use it as a first approximation. Of course, this is not possible in all cases.
Another popular method is to use an effect known as anomalous scattering. This occurs when the X-ray wavelength is close to the electron transition energy in the atoms, which results in the wave being absorbed and re-emitted.
This causes a change in the amplitudes and phases. If the amplitudes are measured very precisely, the differences between them can be used to reconstruct the phases. However, most of the atoms that make up proteins (carbon, oxygen and nitrogen) are not suitable for this. A relatively heavy element found in almost all proteins is sulfur, and this is the element the researchers used in their most recent study to reconstruct the phases.
Special software had to be developed specifically for the task. Out of 7 million images obtained, the researchers had to pick out those with diffracted reflections. They then had to determine the orientation of the crystal and the intensity of all reflections and subsequently collate all the data obtained. 600,000 diffraction patterns were found and then used to successfully reconstruct the structure of a protein with a resolution of 2.5Å. By combining the data with the results obtained at a different X-ray wavelength, the researchers were able to increase the resolution to 1.9Å. This level of precision not only enables the structure of receptor proteins to be determined with high accuracy, but also allows scientists to see molecules of water, ions and lipids that surround them, which is extremely important for understanding how proteins function and modeling their interaction with other substances.
“When I participated in a study to determine the structure of the first receptor protein, it took me about a year to obtain crystals that were large enough to conduct classical X-ray diffraction. We hope that the method we have developed will help to greatly increase the speed of this process,” said Prof. Cherezov commenting on the significance of the research.
Of the 800 receptor proteins that exist, we currently know the structure of only 34. The experimental method developed by the scientists will significantly speed up the studies of the remaining proteins. This, in turn, will help in developing new and effective drugs to treat a vast number of diseases.
 Semaglutide outperforms other GLP-1 drugs for weight loss in psychiatric populations
Semaglutide outperforms other GLP-1 drugs for weight loss in psychiatric populations