The process of expressing a wide variety of voltage-gated and ligand-gated ion channels enables nociceptors to transduce receptor potential into single action potentials or multiple action potentials. As seen in Figure 1, these signals bear the ability to encode the intensity of a noxious stimulus, leading to the transduction of pain signals.
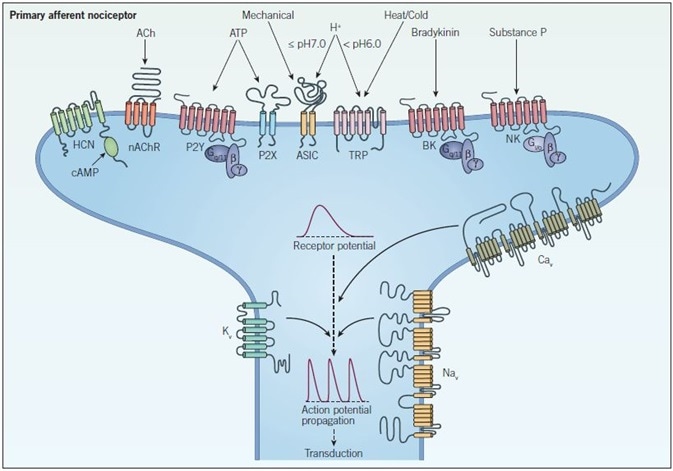
Figure 1. Peripheral sensitization and signal propagation in nociception. Peripheral terminals respond to noxious stimuli through ion channels such as TRP, ASIC, HCN, and P2X receptors and GPCRs such as bradykinin (BK), neurokinin (NK) and P2Y receptors which indirectly modulate ion channels and intracellular signaling pathways. When a threshold depolarization is reached, voltage-gated sodium and calcium channels (NaV and CaV respectively) are activated, which generates an action potential. At this point voltage-gated potassium channels (KV) open and repolarize the membrane, inactivating NaV channels and returning the neuron to a resting state. The action potential then propagates along the axon in a process called transduction.
Transient receptor potential or TRP channels have recently been pursued as pain targets. This is owing to their identification at the genetic level, where they were found to mediate the painful effects of capsaicin (which, in other words, is the chemical responsible for the pungency of chili), via the TRPV1 channel.
From a classical perspective, the understanding of the ion channels involved in pain implicates the sodium channels, since these channels are responsible for determining the excitability of neurons in addition to the calcium channels influencing membrane potential and neurotransmission. Potassium channels play a significant role in repolarizing neurons back to a resting state. This goes hand-in-hand with the emerging roles played by the ASIC, HCN, and P2X receptor ion channels when it comes to pain.
TRP Channels
Ever since the role of TRP channels in pain was discovered, research has largely focused on the identification of compounds by the process of inhibiting their activity, bringing about analgesia. TRP channels, which are named owing to the function they play in phototransduction in Drosophila, can be sub-categorized into six families in mammals – TRPC, TRPM, TRPA1, TRPP, TRPML, and TRPV. Assembling as six transmembrane domains, they are known to generate tetramers of cation-selective channels. Furthermore, each of these possesses different degrees of permeability in terms of calcium and sodium.
Figure 2. TRP channels as temperature sensors. Thermal activation profile of temperature-sensitive TRP channels. Receptor type activated by particular temperatures is highlighted in the lower part of the figure, aligned to a temperature scale bar.
As seen in Figure 2, TRP channels exhibit responsiveness to a range of stimuli – right from temperature, mechanical stress, and alterations in osmolarity, as well as intracellular and extracellular messengers. Further, they are sensitive to voltage in a very weak manner. When opened, TRP channels depolarize cells from the resting membrane potential, raising the concentrations of intracellular sodium and calcium, as well as exciting the cell.
Recent developments – such as the advancement of TRPV1 and TRPV3 channel blockers to clinical trials, as well the advancement of TRPA1 blockers to preclinical development for the treatment of pain – have highlighted the ever-increasing importance of the TRP channel as being a key target in pain. Although all six families play wide and varying roles in several physiological and pathophysiological processes, the TRPV1, TRPM8, and TRPA1 channels are the ones that are researched the most. These cause sensory nerve activation in the DRG, and as a result, they are believed to play an integral role in pain.
Formerly known as the vanilloid receptor, the TRPV1 channel is the most extensively studied among all the TRP channels. It is activated by noxious heat, acidic pH, as well as the pungent extracts that come from chilies, garlic, black pepper, and cinnamon. In several chronic human pain states, TRPV1 expression is increased, which is also the case in many knockout animal models lacking a functional TRPV1 gene that does not exhibit typical responses to painful stimuli.
The importance of TRPV as a novel pain target has been highlighted by the recent generation of selective TRPV blockers – such as the TRPV1-selective blocker JNJ 17203212 as well as the TRPV4-selective blocker RN 1734. This, in turn, has demonstrated significant attenuation of painful symptoms in cancer models. Other TRPV1 blockers – such as capsazepine and A 784168 – have demonstrated the ability to block acute pain that arises as a result of capsaicin and BCTC.
TRPV1 channels possess a multimodal nature, which means that they provide the opportunity to design specific drugs that are modality-specific. Therefore, it is possible to block the activation by distinct stimuli while still ensuring that TRPV1 channels remain sensitive to other stimuli. The feasibility of this approach has been demonstrated by the design of channel blockers that inhibit TRPV1 activation by capsaicin but not acid (e.g. SB 366791), and others that do not differentiate between capsaicin and acid (e.g. AMG 9810).
In the treatment of pain, although most attention is given to the blockage of the TRPV1 channel, paradoxically, the TRPV1 activators such as (E)-capsaicin have also been proven to induce analgesia. For many years, the topical application of TRPV1 activators (e.g. capsaicin creams) has been implemented to alleviate chronic pain conditions such as diabetic neuropathy. This is because it is believed that the desensitization of the capsaicin receptor has the potential to mediate this analgesic effect.
However, it has also been hypothesized that capsaicin is capable of reversibly depleting substance P, which is one of the body's primary neurotransmitters when it comes to pain and heat. The hypothesis further suggests that capsaicin depletes substance P from nerve endings, leading to a reduction in the sensation of pain. As such, resiniferatoxin, i.e. a capsaicin analog, is under evaluation for long-term analgesia, especially in the case of cancer patients with chronic intractable pain.
Furthermore, the TRPA1 channel is the only ankyrin found in mammals. It is predominantly expressed in C-afferent sensory nerve fibers. TRPA1 is also believed to play a vital role in nociception, considering that TRPA1 colocalizes with TRPV1. TRPA1 can be activated by a variety of conditions, such as sub-zero temperatures, mustard oil, cinnamon oil, raw garlic, onions, and formalin. Interestingly, the activation of TRPA1 by these kinds of stimuli elicits a painful burning or prickling sensation.
To combat this effect, a super-cooling agent capable of activating both cold receptors, TRPM8 and TRPA1, is icilin. Icilin is responsible for producing extreme sensations of cold in both humans and animals, with a potency that is almost 200 times stronger than menthol.
Any variation in the gene expression of TRPA1 can significantly alter the perception of pain in humans. Meanwhile, the role played by TRPM8 channel in cold hypersensitivity, analgesia (in neuropathic pain) and inflammation all suggest that it could represent a key therapeutic target. TRPA1 channel blockers such as HC 030031 (Cat. No. 2896) are capable of reducing neuropathic pain and cold hypersensitivity, without altering ordinary cold sensation. Thus, this suggests that such channel blockers are useful tools to understand nociception.
One of the structurally related compounds, i.e. the TCS 5861528, also prohibits the development of mechanical hyperalgesia in animal models where the pain is induced by diabetes. In the rat, for instance, the TRPM8 channel blocker AMTB attenuates the bladder micturition reflex and nociceptive reflex responses and thus has the potential to become a new therapeutic avenue for a variety of syndromes such as overactive bladder and painful bladder. Thus, it is evident that a growing number of TRP channels are of potential therapeutic interest, which in the future could result in novel clinical drugs for pain treatment and therapy.
Sodium Channels
The activity of voltage-gated sodium channels (NaV) is the factor that dictates excitability in neurons. Depolarization activates NaV channels and enables the rapid influx of sodium ions, which generates action potentials. Pain signals, by way of action potentials, are then able to propagate along the axon of the nociceptor by the activation of NaV channels.
In reality, there are nine widely recognized subtypes of NaV channels, which are designated NaV1.1-1.9. Comprised of an alpha subunit that forms four homologous domains (each with six-transmembrane helices), these NaV channels contain two auxiliary beta subunits that are involved in regulation.
Figure 3. Tetraodontidae – a source of tetrodotoxin. The pufferfish (Tetraodontidae) is a well-known source of tetrodotoxin. Although tetrodotoxin was originally discovered in these fish, it is actually produced by symbiotic bacteria that reside within the liver and other organs of the pufferfish.
Largely, as seen in Figure 3, NaV channels are blocked by nanomolar concentrations of tetrodotoxin (TTx), yet NaV1.5, NaV1.8, and NaV1.9 are relatively resistant to TTx (which also goes by the name TTx-R). It has been demonstrated that gain-of-function mutations in NaV channels cause the hyperexcitability of nociceptors. In the adult DRG, most sodium channels, excluding NaV1.4 and NaV1.5, have been identified. However, NaV1.7 and NaV1.8 channels are those that display the greatest influence on nociception.
To support this hypothesis, it has been proven that mice lacking in NaV1.7 and NaV1.8 display deficits in mechanosensation. Conversely, in humans, mutations in the human SCN9A gene that encodes NaV1.7 are associated with three recognized pain disorders, including insensitivity to pain associated with channelopathy, paroxysmal extreme pain disorder as well as primary erythermalgia.
Some of the most useful tools for blocking action potential propagation and excitability in nociceptors are the NaV1.7-selective tarantula venom peptides, ProTx II and Huwentoxin IV. Further, TC-N 1752, which is an orally available blocker of TTx-sensitive sodium channels, is also known to decrease pain sensitization in sensory neurons in the case of rats.
Following the injection of a painful inflammatory agent in rodents, NaV1.8 channel mRNA and protein show an increase in the DRG. In contrast, mechanical allodynia, as well as thermal and mechanical pain hypersensitivity, has been demonstrably reduced in rats by genetic knockdown. NaV1.8 channel blockers such as A 803467 and A 887826 have both been shown to attenuate mechanical allodynia in rat neuropathic pain models.
In the case of most anesthetics, sodium channels and the excitability of all sensory channels are blocked. However, QX 314 bromide – which is a charged lidocaine derivative that would otherwise be impermeant – has been used to selectively target NaV channels by passing through the open pore of a TRPV1 channel. Thus, researchers have been able to produce pain-specific local anesthesia in TRPV1-expressing nociceptors without affecting other sensory neurons through the administration of the TRPV1 agonist (E)-capsaicin in combination with QX 314 bromide. Such a strategy represents an important insight as to how specificity can be created by exploiting ion channels as drug delivery ports.
Acid-Sensing
Pain can sometimes be the outcome of extracellular tissue acidosis that occurs as a result of tissue injury. A drop in pH (i.e. increased acidity) is often identified by ASICs or acid-sensing ion channels in primary sensory neurons (≤pH 7.0) and by TRPV1. Linked to the sodium channel family, ASICs are primarily expressed in central and peripheral neurons, particularly nociceptors, where they are responsible for regulating neuronal sensitivity to acidosis.
Peptides, such as neuropeptide SF, are responsible for regulating the activity of ASICs, which can be blocked by the broad spectrum Na+ channel blocker, amiloride. However, a potent novel ASIC3 specific channel blocker, APETx2, has demonstrated analgesic properties against acid-induced pain. In other words, this channel blocker does not block ASIC1a, 1b or 2a channels. In the future, such newly developed subtype-selective inhibitors will be vital in understanding the role of ASICs in pain.
Calcium Channels
In response to depolarization, voltage-gated calcium channels (CaV) increase intracellular calcium. In turn, this stimulates the release of neurotransmitters such as substance P and CGRP. As a result, this then determines membrane excitability and regulates gene expression. In the propagation and processing of pain signals, these channels play a vital role, with a variety of calcium channels being expressed in nociceptors.
In reality, calcium channels are complex proteins that comprise 4-5 distinct subunits. These subunits are encoded by a variety of different genes. The largest domain is the pore-forming alpha subunit, which, like the sodium channel, comprises four homologous domains. Each of these, in turn, contain six transmembrane helices. This alpha subunit is modulated by subunits such as the auxiliary β, α2δ, and γ, which individually regulate the channel properties. There are five main kinds of calcium channels, which are the L-, P/Q-, N-, R-, and T-type.
In the DRG, studies involving calcium currents were the foundation that led to the functional discovery of the N-type (CaV2.2) calcium channel. Here, the greatest expression was reported at the presynaptic terminal. In mice, knockout studies with N-type (CaV2.2)-null mice demonstrated that these mice bore an increased pain threshold, which demonstrated the involvement of the channels mentioned above in pain. Belonging to a group of conotoxins, the ω-conotoxins are neurotoxic peptides that are isolated from the venom of marine cone snails (Figure 4). In fact, ω-conotoxins are still the most selective inhibitors of N-type calcium channels identified.
Figure 4. Conus textile – a source of conotoxins. The marine cone snail (Conus textile) is a source of the neurotoxic peptide known as conotoxin. Cone snails use a hypodermic-like tooth and a venom gland to attack and paralyze their prey before engulfing it.
In rats, the administration of the N-, P/Qtype calcium channel blocker, i.e. ω-conotoxin MVIIC, reduces pain behavior for a period of up to 24 hours. Thus, researching key tools such as the N-type selective ω-conotoxin GVIA offers the possibility to help unlock the role of these channels in pain transmission. Further, studies of the R-type (CaV2.3) calcium channel through the use of a selective antagonist SNX 482 also demonstrates this channel’s role in chronic neuropathic pain.
In addition, the T-type (CaV3.1-3.3) calcium channel is a channel activated by a low voltage that is expressed in cell bodies and nerve endings of afferent fibers that play a role in the initiation of action potentials. Such T-type channels can lower the action potentials threshold and thus promote bursting and excitation, which in turn can enhance pain sensation. Mibefradil and other T-type channel blockers attenuate hyperalgesia and reverse experimental neuropathic pain.
What is interesting to note is the fact that gabapentin, which is a widely used drug for the treatment of neuropathic pain, and pregabalin both display interaction with the calcium channel α2δ auxiliary subunit. The result is that these reduce channel currents. However, the precise functions of these subunits are not crystal clear, although studies have pointed to their efficacy in the enhancement of channel current. Moreover, peripheral nerve injury has historically demonstrated its ability to upregulate α2δ in the dorsal horn, highlighting their importance as therapeutic targets for neuropathic pain.
Potassium Channels
The principal function of potassium channels is to stabilize the membrane potential. This stabilization is done through the conduction of hyperpolarizing outward potassium ion (K+) currents, which thereby decreases cellular excitability. The ability to re-establish electrical neutrality through potassium channel modulation also enables them to become a potential target that can decrease nociceptor excitability. However, there have been limited studies in this field.
There are four major families of potassium channels: voltage-gated (KV), inward rectifier (Kir), calcium-activated (KCa) and two-pore (K2P). Due to the fact that these channels are diverse in terms of structure and function, they can fulfill a variety of roles in the membrane. Moreover, some of them are also capable of playing a role in antinociception. KV channels’ densities are markedly reduced by peripheral nerve injury, thus implicating them in the development of pain.
Based on their sensitivities to their respective antagonists, the DRG neurons express three distinct classes of K+ currents -- tetraethylammonium (TEA) that block the slow-inactivating sustained K+ current carried by KV7.2/7.3 channels, 4-aminopyridine (4-AP) that block the fast-inactivating transient A-current carried by the KV1.4 channel and α-dendorotoxin (α-DTX) that blocks the slow-inactivating transient D-current carried by KV1.1/1.2 channels.
KV channel openers such as ML 213 display selectivity for a certain channel subtype (KV7.2 and KV7.4) and can be used to identify the role of K+ channel subtypes in pain sensation.
Furthermore, the ATP-sensitive (KATP) channel, which is a member of the Kir family, is inhibited by glibenclamide. This has also been implicated in the pathophysiology of pain. By extension, Kir-selective tools such as tertiapin-Q can also prove useful in the study of pain. However, despite limited understanding of the roles played by potassium channels in pain modulation, they may become promising pain targets in the future.
HCN Channels
HCN channels refer to hyperpolarization-activated cyclic nucleotide-modulated (HCN) channels, which are also prominent in peripheral sensory neurons. HCN channels have been recently proposed as a target for pain treatment as well as allodynia that is associated with nerve injury.
These channels carry an inward current; an unusual characteristic considering that they are activated by membrane hyperpolarization and are modulated by intracellular cAMP. Past evidence suggests that HCN channels may play an important role in the initiation of neuropathic pain, though certain subtypes may also influence the heart rate, reducing their suitability as pain targets. Moreover, the HCN blocker ZD 7288 causes significant suppression of action potential firing in nociceptors, and this is independent of changes in the speed of conduction.
Glutamate (Ionotropic) Receptors
One of the most widely distributed excitatory neurotransmitters in the CNS is glutamate, which is fundamental to excitatory transmission. Emerging evidence supports the notion that modulating glutamate receptors can be potentially used as therapy in the treatment of pain.
Glutamate targets two main types of receptors: the ionotropic receptors, which are ligand-gated ion channels; and the metabotropic receptors, which are coupled to intracellular second messengers through a G-protein signaling cascade. This has been discussed in the next section on GPCRs (Figure 7).
Ligand-gated non-selective cation channels NMDA, AMPA, and kainate receptors are all members of the ionotropic class of glutamate receptors, which, in response to glutamate binding, allow the flow of K+, Na+, and Ca2+. Using animal models, several pharmacological studies have zeroed in on the ionotropic receptors in persistent pain states. In models of first-degree burns, AMPA antagonists such as NBQX and CNQX have demonstrated their effectiveness in blocking the development of hyperalgesia.
In rats and humans alike, blocking the ion channel pore of the NMDA receptor using antagonists such as (+)-MK 801, is effective in alleviating pain. However, targeting different sites on the NMDA receptor on a selective basis can be achieved using specific compounds that permit researchers to block certain aspects of channel activity.
On the NMDA receptor, targeting the alternate glycine modulatory site using the antagonist L-701,324 has so far proven to be an effective method in preclinical animal models for pain. Further, the antagonization of one type of subunit from this receptor, such as the NR2B subunit using Ro 25-6981, has also proven its efficacy in alleviating mechanical allodynia.
Thus, such research not only demonstrates the importance of the ionotropic glutamate receptors in pain but also indicates that selectively targeting subtypes or binding sites within the same receptor has the potential to lead to greater functional specificity in the treatment of pain.
GABAA Receptors
The inhibitory control of nociceptive signals is attributed to γ-aminobutyric acid (GABA). Furthermore, there are two categories of GABA receptors: GABAA and GABAB. GABAC is a third type of GABA receptor that was designated as a receptor. GABAC is insensitive to typical modulators of GABAA. However, it has since been described to be simply a variant within the GABAA receptor family. While GABAA receptors are ligand-gated ion channels or ionotropic receptors, GABAB receptors are G-protein-coupled receptors or metabotropic receptors. These have been discussed in the next section on GPCRs (Figure 7).
Agents were developed owing to the discovery that GABA receptor agonists and inhibitors of GABA uptake or metabolism, were able to display antinociceptive properties. GABAA agonists such as THIP, and the positive allosteric modulator ganaxolone, have all enabled significant analgesia, thus ensuring GABAA receptors are viewed as an interesting target for pain.
P2X Receptors
Receptors in the form of purinergic receptors have been implicated in pain. There are two main types of purinergic receptors: the adenosine receptor (also classed as a P1 receptor) and P2 (ATP) receptor. The latter is further sub-classified into P2X and P2Y receptors (adenosine and P2Y receptors are discussed in the next section on GPCRs). However, the P2X receptor plays a greater role in current pain research streams.
Recent investigations in ATP’s role in the modulation of nociception highlight that there are multiple P2X receptor-based mechanisms through which ATP can facilitate the generation of pain signals. P2X receptors are a family of cation-permeable ligand-gated ion channels. Furthermore, seven subtypes (P2X1-P2X7) of P2X receptors have been found in the central and peripheral mammalian nervous systems.
On an experimental base, P2X receptor agonists such as α,β-Methyleneadenosine 5'-triphosphate, elicit short-lasting nociceptive responses. These are then increased during neuronal sensitization during inflammatory pain. The P2X3 receptor is a potent antagonist TNP-ATP that has been proven to be a useful tool in understanding the role of the P2X3 receptor in vitro. However, due to its rapid degradation, it has limited potential to be used in vivo. Recently, several novel small molecule antagonists that selectively block P2X3 and P2X2/3 receptors, such as the RO-3, Ro 51, and TC-P 262 have been reported –to reduce nociceptive sensitivity in animal models of pain.
In macrophages, microglia, and certain lymphocytes, the P2X7 receptor is highly expressed. Studies into P2X7 selective antagonists have produced substantial evidence supporting their role in animal models of persistent neuropathic and inflammatory pain. Studies that use selective antagonists such as A 438079 and A 740003 have provided direct support for the role of P2X7 receptors in pain modulation. They demonstrate dose-dependent antinociceptive effects in models of neuropathic and inflammatory pain, thus drawing a hypothesis that purinergic glial-neural interactions are important modulators of noxious sensory neurotransmission.
Nicotinic Acetylcholine Receptors
For many years, nicotine has been known to possess weak analgesic activity. Nicotine’s effects are brought about via nicotinic acetylcholine receptors (nAChRs), which are ligand-gated ion channels. These are assembled using a combination of one or more alpha subunits (α1-10), and one or more non-alpha subunits, including β1-4, γ, δ, and ε subunits. It is important to note that chronic injury causes overexpression of key nAChR subunits, such as α4, α5, and α7.
Figure 5. Epipedobates tricolor – a source of epibatidine. Epibatidine is an alkaloid found on the skin of the endangered Ecuadorian frog (Epipedobates tricolor). The frog uses the compound to protect itself from predators, as it can kill animals many times larger than itself.
In several species and pain tests, nicotine and nicotinic agonists-induced activation of cholinergic pathways have been shown to elicit antinociceptive effects. As seen in Figure 5, the nAChR agonist (±)-epibatidine is an alkaloid poison naturally occurring in the skin of the Ecuadorian frog (Epipedobates tricolor). This is approximately 100-200 times more potent than morphine as an analgesic. Despite the fact that it has demonstrated potent antinociceptive effects in neuropathic pain models, it is highly unlikely to be used clinically, owing to intolerable toxic side effects due to non-selectivity.
Thus, the key to developing safe and effective nicotinic agonists as analgesics is primarily to understand which nAChR subtypes are involved in modulating nociceptive transmission. Subsequently, the process will involve developing drugs with increased specificity for these nAChR subtypes. For instance, the effect of nicotine on inhibitory currents could be mimicked by the α4β2 nAChR agonist RJR 2403, and also by choline, an α7 nAChR agonist. However, while the effect of nicotine can potentially be completely blocked by the nAChR antagonist mecamylamine and the α4β2 nAChR antagonist dihydro-β-erythroidine, these display differing consequences on nAChR activity. In the sensation of pain, α4β2 agonists such as sazetidine A, the partial agonist varenicline, as well as the α7 agonist LY 2087101 will help to elucidate the role played by different receptor subtypes.
Thus, in studies investigating acute pain, modulators of the α7 receptor such as the antagonists, methyllycaconitine and α-bungarotoxin, and the agonists, PNU 120596, AR-R 17779 and A 844606, have proven to be useful. This further suggests that subtype specificity may hold key answers in understanding the role of nicotinic receptors in pain.
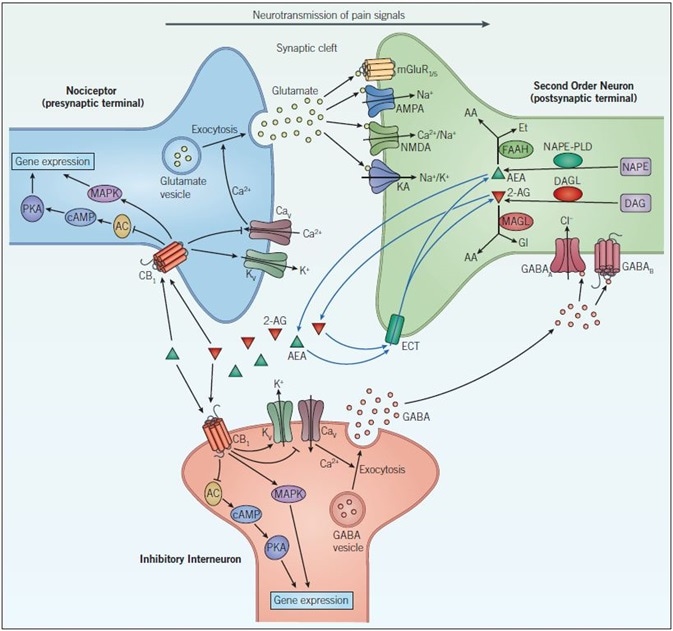
Figure 7. Endocannabinoid signaling and neurotransmission. Pain signals from the nociceptor arrive at the presynaptic terminal whereupon opening of voltage-gated calcium channels (CaV), elevates intracellular calcium, and stimulates exocytosis so that glutamatergic signaling can occur. Glutamate acts on two different types of receptors on the postsynaptic neuron: ionotropic glutamate receptors (NMDA, AMPA, and KA) and metabotropic glutamate receptors (mGluR). Activation of these channels and receptors initiates a receptor potential in the postsynaptic neuron. Endocannabinoids (such as anandamide (AEA) and 2-arachidonoylglycerol (2-AG)) are synthesized in response to increased activity in the postsynaptic neuron. They exert their effects by binding to specific G-protein-coupled receptors located on the presynaptic neuron. The CB1 receptor inhibits the AC-cAMP‑PKA pathway and activates the mitogen-activated protein kinase (MAPK) cascade, both of which regulate gene expression. The CB1 receptor modulates ion conductances, inhibiting voltage-sensitive Ca2+ channels, which blocks exocytosis and activates voltage-sensitive K+ channels (KV), leading to suppression of the signals coming from the nociceptor – this is called retrograde signaling. Endogenous cannabinoids in the synaptic cleft are taken up by endogenous cannabinoid transporters (ECT) into the cell whereby they are broken down by enzymes that include, fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL). Inactivation of AEA (by FAAH) and 2-AG (by MAGL) occurs via hydrolysis to arachidonic acid (AA) and ethanolamine (Et) or glycerol (Gl), respectively. GABAergic neurons also act on the postsynaptic cell to decrease excitability. GABAA, a ligand-gated ion channel that conducts chloride ions and GABAB, a G-protein-coupled receptor that is linked to K+ channels and that inhibits the AC-cAMP-PKA pathway, leads to inhibition of the postsynaptic neuron and a dampening down of pain signals communicated to the brain.
About Tocris Bioscience
Tocris Bioscience is your trusted supplier of high-performance life science reagents, including receptor agonists & antagonists, enzyme inhibitors, ion channel modulators, fluorescent probes & dyes, and compound libraries. Our catalog consists of over 4,500 research tools, covering over 400 protein targets enabling you to investigate and modulate the activity of numerous signaling pathways and physiological processes.
We have been working with scientists for over 30 years to provide the life science community with research standards, as well as novel and innovative research tools. We understand the need for researchers to trust their research reagents, which is why we are committed to supplying our customers with the highest quality products available, so you can publish with confidence.
Tocris is part of the protein sciences division of Bio-Techne, which also includes the best in class brands R&D Systems, Novus Biologicals, ProteinSimple, and Advanced Cell Diagnostics. Bio-Techne has united these brands to provide researchers with a full portfolio of research reagents, assays, and protein platforms. For more information on Bio-Techne and its brands, please visit bio-techne.com.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices, and treatments.