Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease. Its characteristic features include mental deterioration, psychiatric disorder and motor dysfunction, primarily chorea, an involuntary movement disorder.
It is believed that these symptoms stem from the cellular aggregation and activity of mutant huntingtin protein (Htt) in the cytoplasm and nuclei of neurons. While Htt is expressed across the central nervous system, medium spiny neurons (MSNs) in the basal ganglia of the striatum display specific susceptibility to neurodegeneration in HD.
At present, there are no disease-modifying treatments available for HD, with the only current FDA-approved drug for HD being tetrabenazine, a reversible inhibitor of vesicular monoamine transporter type 2 (VMAT2). Present research is aimed at the identification of novel therapeutic targets, several of which are outlined below.
Etiology of Huntington’s Disease
Htt is primarily a cytoplasmic protein that is essential for embryogenesis and development. It is believed to be connected to a range of processes, including protein trafficking and vesicle transport. As such, a possible role as a scaffold protein is plausible.
Htt is encoded by the IT15 gene, in which the unstable expansion of a three-base sequence (CAG, encoding glutamine) in exon 1 leads to the production of a protein with a lengthened stretch of glutamine residues (a polyglutamine (polyQ) tract) at its amino terminus.
If more than 35 CAG recurrences are found, the protein expressed (mutant huntingtin, mHtt) has characteristics that vary from the standard protein product. Both mutant and wild-type (wt) alleles are expressed in HD. If there are more than 40 CAG repeat elements in an allele, then it is genetically penetrant.
Domains containing multiple glutamine residues are frequently linked to the mediation of protein-protein interactions and require a particular level of inherent conformational flexibility.
Mutations with have an impact on this area are thought to affect this flexibility and subsequently, protein activity. mHtt, and fragments of it, stimulate both deleterious and compensatory processes that leave affected neurons more vulnerable to general injuries, such as oxidative and excitotoxic stressors.
Several transgenic in vivo and in vitro models have been produced to study the pathology and behavioral phenotypes of HD, with specific focus on motor deficits, intranuclear inclusions, transcriptional dysregulation, mitochondrial dysfunction and excitotoxicity.
Employing these models as a foundation for research into the processes underlying the motor and nonmotor symptoms of HD has been successful; however, presently available treatments for HD have not been able to offset the advancement of the disorder.
Nonetheless, treatment of the movement and behavioral disorders linked to HD may alleviate disease symptoms, while regenerative therapies could supplant impaired neurons. Figure 1 offers a summary of therapeutic interventions in HD.
Proteolysis and Inclusion Bodies
Inclusion bodies are a pathological symbol of HD and are typically seen in cells where mHtt is expressed. They are made up of protein aggregates containing toxic N-terminal mHtt fragments, which are produced by the caspase, calpain and matrix metalloproteinase (MMP) enzymes that are expressed in MSNs.
Aggregates also include various sequestered proteins, including ubiquitin and chaperone proteins. Nuclear aggregates are mostly made up of the deadly mHtt fragments, while cytoplasmic inclusions contain both full-length and truncated mHtt.
Whether inclusion bodies simply coincide with disease or if they themselves are cytotoxic is not entirely known. Several lines of evidence indicate that by isolating diffuse mHtt fragments, inclusions extend cell survival.
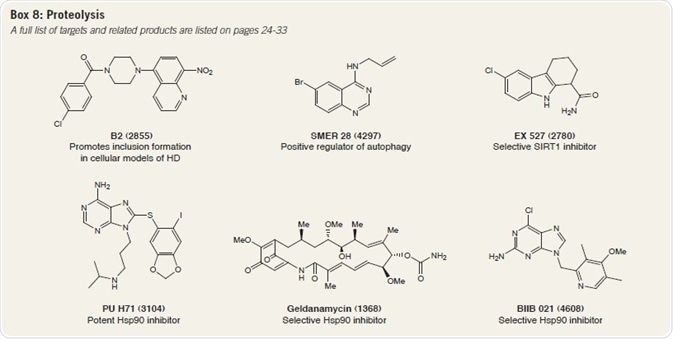
As an example, the small molecule B2 is proven to encourage inclusion formation, while also inhibiting mHtt-mediated proteasome dysfunction in a cellular HD model. B2-induced inclusion formation also leads to a reduction in α-synuclein toxicity in Parkinson's disease (PD).
Enlightenment on disease progression may, therefore, be obtained through greater a understanding of the mechanisms behind mHtt processing and inclusion body formation. It has also been suggested that the presence of cellular aggregates fuels autophagy.
Conversely, prevention of autophagy increases aggregate formation and soluble mHtt levels.

Figure 1. Therapeutic interventions in Huntington’s disease. Huntingtin protein synthesis and processing are integral to HD pathology. mHtt is generated by translation of an allele containing over 35 CAG repeats. It is processed by proteolytic enzymes (namely calpains, caspases and MMPs) to generate toxic N-terminal fragments. These fragments form inclusion bodies in the cytoplasm and nucleus of the neuron, a key hallmark of HD pathology. The interactions of mHtt with a variety of cellular processes and proteins has resulted in the identification of numerous therapeutic targets; a range of small molecules acting at these targets exhibit beneficial effects in transgenic HD models. These include: antioxidants; calcium signaling inhibitors; protease inhibitors; autophagy activators; SIRT1 inhibitors; inducers of heat shock response; histone deacetylase (HDAC) inhibitors; promoters of inclusion formation; and finally, small interfering RNA (siRNA) (examples shown in green). Small molecules have also been used to generate toxin models of HD (examples shown in red). Abbreviations: 3-NP – 3-Nitropropionic acid; BDNF – brain-derived neurotrophic factor; Hsp40 – heat shock protein 40; Hsp70 – heat shock protein 70; IP3R – IP3 receptor; MMPs – matrix metalloproteinases; NMDAR – NMDA receptor; QA – quinolinic acid; ROS – reactive oxygen species; UPS – ubiquitin-proteasome system; VGCC – voltage-gated calcium channel.
A range of autophagy-inducing compounds have demonstrated effectiveness in HD models: rapamycin suppresses mTOR, a negative regulator of autophagy that is also sequestered into aggregates.
Rapamycin has been proven to reduce Htt accumulation in cell models, and also defends against neurodegeneration in a fly model of HD. SMER 28 boosts autophagosome synthesis and improves the clearance of mHtt in mammalian cells.
Furthermore, the Ca2+ channel blocker and autophagy activator, verapamil, demonstrates neuroprotective properties in a range of HD models.
The deacetylase sirtuin 1 (SIRT1) inhibits the autophagic degradation of Htt by eliminating its acetyl tags. As such, selective SIRT1 inhibitors, such as EX 527, may support Htt degradation through autophagy and boost its clearance.
However, deacetylation of regulatory transcription factors by SIRT1 also encourages the expression of neuroprotective genes and brain-derived neurotrophic factor (BDNF).
As a result of its abnormally long polyglutamine tract, mHtt is prone to misfolding. Since chaperone proteins can re-fold misfolded proteins and can subdue the production of polyglutamine oligomers that later form aggregates, they are of key interest in HD research.
Chaperones are also known as heat shock proteins, and the three of these which hold particular significance in HD are Hsp90, Hsp70 and Hsp40. Hsp70 and Hsp40 have been proven to diminish the formation of fibrils by polyglutamine proteins, while Hsp90 lists Htt and mHtt amongst its client proteins.
Suppression of Hsp90 destabilizes huntingtin, assisting in its clearance by the ubiquitin-proteasome system (UPS). Furthermore, Hsp90 suppression, by compounds such as geldanamycin, causes the expression of Hsp70 and Hsp40, constraining mHtt aggregation by triggering a heat shock response.
Celastrol also triggers a heat shock response and has been acknowledged in various screens for Htt aggregation. Further Hsp90 inhibitors, such as PU H71 and BIIB 021 may also be valuable tools (Box 8).
Transcriptional Dysregulation
mHtt and its fragments can bind directly to transcription factors, modifying gene expression. Intranuclear aggregates containing mHtt sequester CBP (CREB-binding protein), the protein that binds CREB (cyclic AMP response element-binding protein).
mHtt binds to the acetyltransferase domain of CBP, as well as the co-activator p300. The subsequent decrease in acetyltransferase activity can be counteracted by use of histone deacetylase (HDAC) inhibitors; sodium butyrate increases chances of survival, while brain-penetrant SAHA diminishes motor deficits in HD mouse models.
Transcriptional dysregulation has an impact on levels of BDNF and its receptor, TrkB, both of which are frequently downregulated in HD. Lowering BDNF production causes a rise in neuronal loss, so returning it to usual levels could assist in the survival of striatal neurons.
Boosting BDNF levels by upstream and downstream pathways may be therapeutically feasible options. Infusion of recombinant BDNF, use of cell grafts releasing BDNF, and BDNF mimetics are all possible approaches currently going through foundational research.
TrkB receptors may be transactivated by adenosine A2A receptors; suppression of the latter, by compounds such as SCH 58261, is neuroprotective in excitotoxic models of HD, indicative of a role for TrkB in excitotoxicity.
Mitochondrial Dysfunction
Neurons typically need high levels of ATP to uphold functions such as membrane polarization and vesicle trafficking. In particular, MSNs need large quantities of energy from oxidative phosphorylation, and are thus, particularly sensitive to the mitochondrial dysfunction that is often manifest in HD models.
mHtt associates with the outer membrane of mitochondria, leading to damage to the electron transport chain complexes II and III.
Succinate dehydrogenase (mitochondrial complex II), can be targeted by the compounds 3-Nitropropionic acid (3-NP) and methylmalonate; 3-NP causes HD disease-like pathology, while methylmalonate brings about neuronal cell death.
Diminished mitochondrial function also has proven links with greater activity of the enzyme transglutaminase 2 (TG2). TG2 accrues in cells under stress and is upregulated in animal models of HD. Its erasure in two HD mouse models resulted in better survival rates and lowered neuronal death.
Pharmacological suppression of TG2 reverses the vulnerability of human HD cells to 3-NP and also alleviates transcriptional dysregulation. Cystamine is a transglutaminase inhibitor that also boosts BDNF secretion (and therefore, endogenous BDNF levels). It has been proven to boost motor performance and extend survival in HD mice.
Mitochondria share close links with the generation of reactive oxygen species (ROS). Excess levels of ROS lead to oxidative stress, which is heightened in HD cells. As such, antioxidants have been studied in HD trials. Nrf2, a transcription factor linked to the antioxidant response, and which is triggered by stress, is also neuroprotective in an HD model.
Nrf2 decreases the mean lifetime of polypeptides containing a polyQ expansion, and additionally, boosts neuronal survival in vitro. As a result, Nrf2 is a crucial contributor to mHtt clearance and activators of Nrf2, such as TAT 14, DMF and MMF may be beneficial tools.
Excitotoxicity
Glutamate
As outlined above, HD predominantly has an impact on striatal MSNs, a population of neurons that receives both glutamate signals from the cortex and dopamine signals from the substantia nigra.
An established hypothesis suggests that high levels of excitatory neurotransmitters and/or activation of postsynaptic glutamate receptors (particularly the NMDA receptor) on MSN membranes sensitizes them to excitotoxic cell death.
The grouping of various glutamatergic afferents and distinctive NMDA receptor subtype composition (NR1A and NR2B) in MSNs could cause them to be susceptible to harm in HD. Since MSNs are GABAergic, the forfeiture of their inhibitory input to the globus pallidus is believed to be an underlying cause of the choreic movements typical of HD.
mHtt is thought to increase the activity of NMDA receptors, and NR1A and NR2B are especially vulnerable to an increase in current flow mediated by mHtt. Agonists and antagonists of NMDA receptors have been widely employed in fundamental HD research.
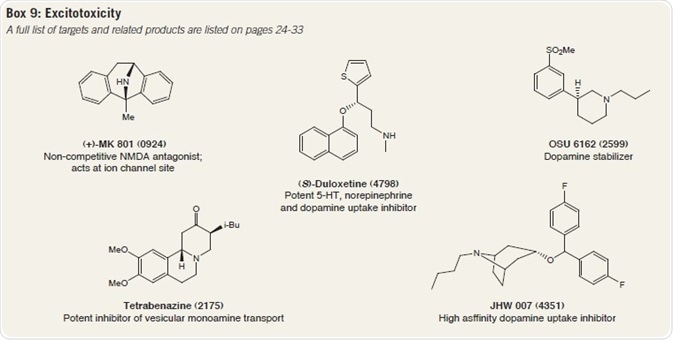
Quinolinic acid, an endogenous NMDA receptor agonist, is frequently employed to produce HD models, while the NMDA receptor antagonist (+)-MK-801 has been proven to inhibit neuronal cell loss brought about by mitochondrial toxins.
Dimebon displays affinity for NMDA receptors and shows neuroprotective effects in HD cellular models, but its clinical development for HD therapy has ceased (Box 9).
Rate of glutamate uptake has been found to have an inverse relation to the number of CAG repeats; rate of glutamate uptake, and thus, clearance from the synaptic cleft, is low in HD brain tissues.
The expression and function of the rodent glutamate transporter GLT-1 (ortholog of EAAT2) are diminished by mHtt. Therefore, boosting glutamate uptake or preventing its release may be beneficial. The anticonvulsant lamotrigine impedes glutamate release and may slow the advancement of HD.
However, the glutamate release inhibitor riluzole failed to demonstrate any advantageous effects in a phase III HD trial.
Dopamine
Dopamine is believed to act in synergy with glutamate to boost Ca2+ levels, sensitizing striatal neurons to mHtt toxicity and inducing apoptosis in MSNs.
High doses of dopamine can also prompt cell death of striatal neurons, most probably through oxidative stress. Continuously administering the dopamine precursor L-DOPA causes a rise in dopamine levels and increases loss of MSNs in HD mice, as well as aggravating dyskinetic symptoms in HD patients.
On the other hand, L-DOPA has a favorable outcome for PD patients, returning motor function by boosting dopamine concentrations that are affected by degeneration of the substantia nigra. Motor dysfunction and MSN loss are also apparent in dopamine transporter knockout mice, which have dependably high dopamine levels.
Dopamine ‘stabilizers’, such as pridopidine and OSU-6162, have been found to reduce locomotor activity in animal models when dopamine levels are high. Both bind and quickly detach from dopamine D2 receptors, which, together with D1 receptors, are broadly expressed in MSNs.
Contemporary research has indicated that the impacts of these compounds may also be mediated by σ1 receptors, for which they demonstrate nanomolar affinity in vitro.
Tetrabenazine, the reversible VMAT2 inhibitor, is thought to augment motor function by draining dopamine storage vesicles. However, it displays some severe side-effects such as depression as a result of lowered serotonin levels.
A further VMAT2 inhibitor, reserpine, also reduces dopamine stores, but displays irreversible binding and is more deadly.
Stem Cells
Stem cells have uses in several diseases, particularly in neurodegenerative disorders. The three key types of stem cells are embryonic stem cells (ESCs), induced pluripotent stem cells (iPSCs) and adult stem cells, the latter of which include mesenchymal, hematopoietic and neural stem cells.
Since they can be derived from easily acquired somatic cells that contain the disease mutation, iPSCs are of great interest in relation to HD research.
They offer researchers a source of cells for drug screening, disease modeling and cell replacement therapy, following directed differentiation into the cell type of interest (e.g. medium spiny neurons). Neuronal distinction of stem cells can be attained with a variety of small molecules (Box 10).
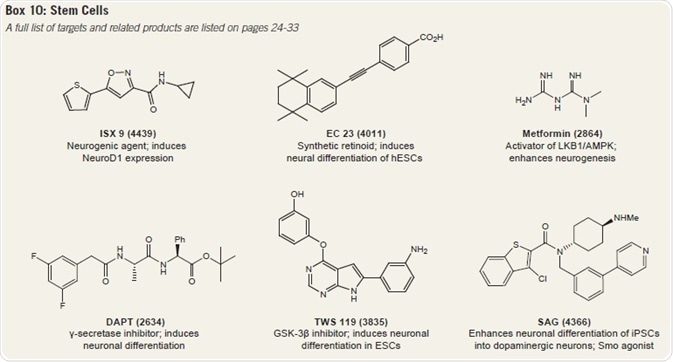
The synthetic retinoid EC 23 and GSK-3β inhibitor TWS 119 can both bring about neuronal differentiation in ESCs, while the γ-secretase inhibitor DAPT causes neuronal differentiation from ESC-derived embryoid bodies.
SAG is a hedgehog signaling activator that prompts the differentiation of dopaminergic neurons from iPSCs.
Other compounds include ISX 9, which boosts NeuroD1 expression to cause cortical neuron differentiation, and metformin, which stimulates neurogenesis from neural precursors.
Varied stem and progenitor cells can, therefore, be lured into precise cell types for regenerative therapy. Contemporary research has produced replacement MSNs from neural stem cells through use of BDNF and noggin proteins.
Transplantation of ESC-derived GABA neurons was found to rectify motor problems in quinolinic acid-lesioned mice, while transplantation of human adipose-derived stem cells was also found to lessen the loss of striatal neurons and reduce the number of Htt aggregates in a quinolinic acid-lesioned rat model.
While ex vivo techniques (i.e. drug screening) may be openly realizable, the transplantation of modified cells into humans is bound by immense scrutiny and legislature, since the safety of such therapies is unclear.
Similarly, the purpose and integration of these cells would need to be evaluated. Nonetheless, stem cells offer a potentially workable alternative to animal or in vitro models of HD, which do not always repeat the disease characteristics exactly.
Additional Targets
Kynurenine Monooxygenase
The enzyme kynurenine 3-monooxygenase (KMO, also known as kynurenine 3-hydroxylase) is linked to tryptophan metabolism. Metabolites in the kynurenine pathway, produced through tryptophan degradation, are associated with HD. Excitotoxicity and free radical production are linked to low levels of kynurenic acid.
The KMO inhibitor Ro 61-8048 boosts levels of kynurenic acid, diminishing levels of extracellular glutamate and inhibiting synaptic loss in a mouse model of HD. Greater concentrations of kynurenic acid also provoke the glycine site of NMDA receptors.
ERK Pathway
The antioxidants fisetin (Figure 2) and resveratrol, have been proven to enhance cell survival rates and display neuroprotective activity, respectively, in two distinct HD models. This activity is believed to stem from the activation of ERK by these two compounds.
Fisetin has also been found to lessen neurodegeneration in flies, and boost the median lifespan of flies and mice expressing mHtt. Furthermore, ERK may be triggered by TrkB receptors after BDNF binding.
In Drosophila glial cells, mHtt impedes ERKdependent expression of glutamate transporters, which could add to excitotoxicity. Therefore, ERK inhibitors, such as FR 180204 and TCS ERK 11e may be worthwhile tools for examining the role of ERK in HD pathology.
Psychiatric Symptoms
Antidepressants (chiefly selective serotonin reuptake inhibitors, SSRIs) and antipsychotics may also be used to counteract the psychiatric expressions of HD. SSRIs, such as citalopram, boost serotonin levels by inhibiting its reuptake into presynaptic cells. In doing so, they increase BDNF levels.
Specific SSRIs have been proven to boost neurogenesis, motor control and cognitive ability in mouse HD models.
As an example, paroxetine mitigates motor dysfunction and boosts survival in HD mice while fluoxetine has been found to increase cognitive function in transgenic HD mice, as well as enhancing neurogenesis by boosting neuronal differentiation of proliferating cells.
Sertraline also supports neurogenesis and increases BDNF levels in R6/2 transgenic mice.

Figure 2. Vaccinium corymbosum – a source of fisetin.
Future Directions
Although there are no existing therapies able to halt or reverse the advancement of HD, it is hoped that the unearthing and/or clarification of possible drug targets may help spur on the development of disease-modifying therapies.
Although they demonstrate similar neurodegenerative mechanisms, HD is distinct from AD and PD in that it is an entirely genetic disorder.
Subsequently, gene silencing is a current area of research with HD mouse models, with small interfering RNAs (siRNAs) and antisense oligonucleotides being employed to selectively target the messenger RNA (mRNA) encoded by the mutant allele. Clinical trials in HD patients employing this method will potentially take place in the near future.
As is the case with other neurodegenerative diseases, stem cells may be advantageous in the future. These can be coaxed into particular cell types for regenerative therapy, hopefully replacing the lost neurons and returning normal brain function.
Central questions relating to the roles of Htt and its connections with intracellular proteins are currently unanswered, but it is hoped that investigation of Huntington’s disease may help inform research into other neurodegenerative disorders with comparable pathologies.
Further Reading
Please refer to the list of recommended papers for more information.
Alzheimer’s Disease
- De Strooper et al (2010) The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 6 99
- Eckert et al (2012) Mitochondrial dysfunction-a pharmacological target in Alzheimer’s disease. Mol. Neurobiol. 46 136
- Francis et al (1999) The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J. Neurol. Neurosurg. Psychiatry
- 66 137
- Geldenhuys et al (2011) Role of serotonin in Alzheimer’s disease: a new therapeutic target? CNS Drugs 25 765
- Goedert et al (1991) Tau proteins and neurofibrillary degeneration. Brain Pathol. 1 279
- Goncalves et al (2013) Amyloid β inhibits retinoic acid synthesis exacerbating Alzheimer disease pathology which can be attenuated by an retinoic acid receptor α agonist. Eur. J. Neurosci. 37 1182
- Hynd et al (2004) Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 45 583
- LaFerla et al (2007) Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 8 499
- Massoud and Gauthier (2010) Update on the pharmacological treatment of Alzheimer’s disease. Curr. Neuropharmacol. 8 69
- Medhurst et al (2009) Characterization of histamine H3 receptors in Alzheimer’s Disease brain and amyloid over-expressing TASTPM mice. Br. J. Pharmacol. 157 130
- Noble et al (2013) The importance of tau phosphorylation for neurodegenerative diseases. Front Neurol. 4 83
- Ramírez et al (2005) Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J. Neurosci. 25 1904
- Salomone et al (2012) New pharmacological strategies for treatment of Alzheimer’s disease: focus on disease modifying drugs. Br. J. Clin. Pharmacol. 73 504
- Savelieff et al (2013) Untangling amyloid-β, tau, and metals in Alzheimer’s disease. ACS Chem. Biol. 8 856
- Selenica et al (2007) Efficacy of small-molecule glycogen synthase kinase-3 inhibitors in the postnatal rat model of tau hyperphosphorylation. Br. J. Pharmacol. 152 959
- Serý et al (2013) Molecular mechanisms of neuropathological changes in Alzheimer’s disease: a review. Folia Neuropathol. 51 1
- Siedlak et al (2009) Chronic antioxidant therapy reduces oxidative stress in a mouse model of Alzheimer’s disease. Free Radic. Res. 43 156
Parkinson’s Disease
- Abou-Sleiman et al (2006) Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat. Rev. Neurosci. 7 207
- Exner et al (2012) Mitochondrial dysfunction in Parkinson’s disease: molecular mechanisms and pathophysiological consequences. EMBO J. 31 3038
- Henchcliffe and Beal (2008) Mitochondrial biology and oxidative stress in Parkinson’s disease pathogenesis. Nat. Clin. Pract. Neurol. 4 600
- Huot et al (2013) The pharmacology of L-DOPA-induced dyskinesia in Parkinson’s disease. Pharmacol. Rev. 65 171
- Lanciego et al (2012) Functional neuroanatomy of the basal ganglia. Cold Spring Harb. Perspect. Med. 2 a009621
- Lee et al (2012) Apoptosis signal-regulating kinase 1 mediates MPTP toxicity and regulates glial activation. PLoS One. 7 e29935
- Lee et al (2013) Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat. Neurosci. 16 1392
- Lundblad et al (2012) Impaired neurotransmission caused by overexpression of α‑synuclein in nigral dopamine neurons. Proc. Natl. Acad. Sci. U. S. A. 109 3213
- McNaught et al (2003) Altered proteasomal function in sporadic Parkinson’s disease. Exp. Neurol. 179 38
- Surmeier and Schumacker (2013) Calcium, bioenergetics, and neuronal vulnerability in Parkinson’s disease. J. Biol. Chem. 288 10736
- Venderova and Park (2012) Programmed cell death in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2 a009365
Huntington’s Disease
- Arraste et al (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431 805
- Bano et al (2011) Neurodegenerative processes in Huntington’s disease. Cell Death Dis. 2 e228
- Beal and Ferrante (2004) Experimental therapeutics in transgenic mouse models of Huntington’s disease. Nat. Rev. Neurosci. 5 373
- Crook and Housman (2011) Huntington’s disease: can mice lead the way to treatment? Neuron 69 423
- Frank and Jankovic (2010) Advances in the pharmacological management of Huntington’s disease. Drugs 70 56
- Kaplan and Stockwell (2012) Therapeutic approaches to preventing cell death in Huntington disease. Prog. Neurobiol. 99 262
- Labbadia and Morimoto (2013) Huntington’s disease: underlying molecular mechanisms and emerging concepts. Trends Biochem. Sci. 38 378
- Landles and Bates (2004) Huntingtin and the molecular pathogenesis of Huntington’s disease. Fourth in Molecular Medicine Review Series. EMBO Rep. 5 958
- Perrier and Peschanski (2013) How can human pluripotent stem cells help decipher and cure Huntington’s disease? Cell Stem Cell 11 153
- Ravikumar et al (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36 585
- Ross and Tabrizi (2011) Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 10 83
About Tocris Bioscience
Tocris Bioscience is your trusted supplier of high-performance life science reagents, including receptor agonists & antagonists, enzyme inhibitors, ion channel modulators, fluorescent probes & dyes, and compound libraries. Our catalog consists of over 4,500 research tools, covering over 400 protein targets enabling you to investigate and modulate the activity of numerous signaling pathways and physiological processes.
We have been working with scientists for over 30 years to provide the life science community with research standards, as well as novel and innovative research tools. We understand the need for researchers to trust their research reagents, which is why we are committed to supplying our customers with the highest quality products available, so you can publish with confidence.
Tocris is part of the protein sciences division of Bio-Techne, which also includes the best in class brands R&D Systems, Novus Biologicals, ProteinSimple, and Advanced Cell Diagnostics. Bio-Techne has united these brands to provide researchers with a full portfolio of research reagents, assays, and protein platforms. For more information on Bio-Techne and its brands, please visit bio-techne.com.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices, and treatments.