In nociception, a range of G-Protein-Coupled Receptors (GPCRs) plays extremely important roles. By modulating the function of a wide variety of ion channels, GPCRs also signal molecules in the case of sensory neurons.
G-protein activation and inactivation has a basic cycle, which involves agonist binding. This is in addition to receptor activation. In turn, this induces a conformational change in such a way that the α-subunit binds GTP in exchange for GDP. Thus, this causes it to dissociate into a GTP-bound α-subunit and a βγ dimer.
There are several different classes of Gα subunits such as Gαs (G stimulatory), Gαi (G inhibitory), Gαo (G other), Gαq/11, and Gα12/13. These have the capacity to signal to downstream targets to induce the stimulation or inhibition of cellular activity. The inactivation of the G-protein occurs due to the hydrolysis of GTP, thus returning it to its heteromeric state.
Cannabinoids have become a new and exciting area of pain research. Today, research in this area builds upon the knowledge that has been derived from the investigation into the more traditional targets such as the opioid, glutamate, and GABA receptors. Such research has greatly expanded the field of GPCRs in nociception. Also playing a key role in nociception are bradykinin and neurokinin receptors, which are responsible for mediating neurotransmission in pain pathways, making them useful targets for current research.
Cannabinoid Receptors
For centuries, cannabinoids’ analgesic properties have been recognized. Their mechanism of action has only been understood since the characterization of the metabotropic cannabinoid receptors – CB1 and CB2. These receptors inhibit adenylyl cyclase by coupling Gαi/o. The CB1 receptor is also responsible for modulating calcium and potassium conductances and activating mitogen-activated protein kinase (MAPK). This leads to inhibition of cellular activity and suppression of neuronal excitability (Figure 1).
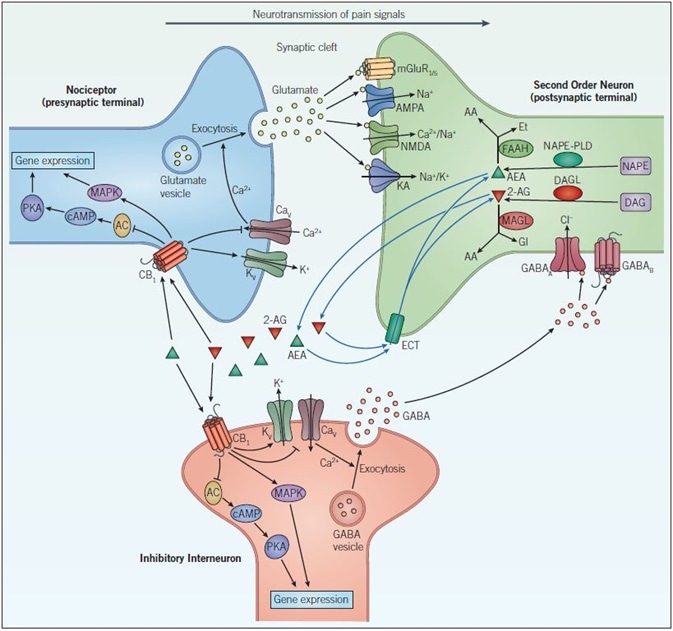
Figure 1. Endocannabinoid signaling and neurotransmission. Pain signals from the nociceptor arrive at the presynaptic terminal whereupon the opening of voltage-gated calcium channels (CaV), elevates intracellular calcium, and stimulates exocytosis so that glutamatergic signaling can occur. Glutamate acts on two different types of receptors on the postsynaptic neuron: ionotropic glutamate receptors (NMDA, AMPA and KA) and metabotropic glutamate receptors (mGluR). Activation of these channels and receptors initiates a receptor potential in the postsynaptic neuron. Endocannabinoids (such as anandamide (AEA) and 2-arachidonoylglycerol (2-AG)) are synthesized in response to increased activity in the postsynaptic neuron. They exert their effects by binding to specific G-protein-coupled receptors located on the presynaptic neuron. The CB1 receptor inhibits the AC-cAMP‑PKA pathway and activates the mitogen-activated protein kinase (MAPK) cascade, both of which regulate gene expression. The CB1 receptor modulates ion conductances, inhibiting voltage-sensitive Ca2+ channels, which blocks exocytosis and activates voltage-sensitive K+ channels (KV), leading to suppression of the signals coming from the nociceptor – this is called retrograde signaling. Endogenous cannabinoids in the synaptic cleft are taken up by endogenous cannabinoid transporters (ECT) into the cell whereby they are broken down by enzymes that include, fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL). Inactivation of AEA (by FAAH) and 2-AG (by MAGL) occurs via hydrolysis to arachidonic acid (AA) and ethanolamine (Et) or glycerol (Gl), respectively. GABAergic neurons also act on the postsynaptic cell to decrease excitability. GABAA, a ligand-gated ion channel that conducts chloride ions and GABAB, a G-protein-coupled receptor that is linked to K+ channels and that inhibits the AC-cAMP-PKA pathway, leads to inhibition of the postsynaptic neuron and a dampening down of pain signals communicated to the brain.
In contrast, the CB2 receptor seemingly does not gate ion channels; however, it does activate MAPK. In addition, an orphan G-protein-coupled receptor, GPR55, has been recently described to bind cannabinoids.
N-acyl ethanolamines (NAE) and monoacylglycerols (MAGL) are two major classes of lipids that activate cannabinoid receptors. Anandamide (AEA), which is an NAE, was the first endocannabinoid to be isolated from the brain, which was closely followed by the monoacylglycerol, 2-Arachidonylglycerol (2-AG).
Further, AEA is synthesized from N-arachidonoyl phosphatidylethanolamine (NAPE) via multiple pathways – including enzymes such as phospholipase A2, phospholipase C, and NAPE-PLD. Through the action of diacylglycerol lipase (DAGL), arachidonic acid-containing diacylglycerol (DAG) is what synthesizes 2-AG. Since then, several other putative endocannabinoids have been found, which include noladin ether, virodhamine, and NADA.
Moreover, the endocannabinoids are believed to be synthesized on demand by activity-dependent or receptor-stimulated cleavage of membrane lipid precursors. These are then released from postsynaptic cells immediately following their production. Once this occurs, the endocannabinoids are then able to regulate the release of a neurotransmitter on the presynaptic neuron through CB receptors, which influences calcium influx driving exocytosis. Thus, as seen in Figure 1, they can control GABAergic or glutamatergic transmission by retrograde signaling.
In the CNS mainly, the CB1 receptor is found, which has been identified at the peripheral and central terminals of primary afferents. In states of neuropathic pain, the levels of CB1 protein and mRNA are increased in DRG neurons projecting into the injured nerve. Conversely, CB1 knockout studies have shown how, in mice, these are hypersensitive to mechanical stimulation.
Furthermore, agonists for the CB1 receptor such as ACEA and WIN 55,212-2 have displayed their efficacy in alleviating mechanical allodynia, acute pain, and hyperalgesia in vivo, through both peripheral and central mechanisms. Moreover, in the spinal cord, the non-selective cannabinoid receptor agonist CP 55,940 has exhibited antinociceptive effects, and application of the selective CB1 agonist ACEA has inhibited spinal nociceptive transmission.
However, the association of cannabinoids with psychotropic side effects, temporary memory impairment, and dependence is the main hurdle in activating CB1 receptors in the CNS for the alleviation of pain. These side effects arise as an effect on forebrain circuits. Therefore, the clinical exploitation of cannabinoids needs to primarily overcome their adverse side effects, if it is to harness its analgesic properties.
Originally, the CB2 receptor was thought to be localized solely to the immune system. However, it has now also been identified in the brain, DRG, spinal cord, and sensory neurons. The CB2 agonists HU 308, JWH 133, and GW 405833 have directly supported the hypothesis that CB2 produces antinociceptive effects in persistent pain states. More importantly, CB2 selective agonists lack the centrally mediated side effects on motility, body temperature, or cognition that are typically associated with CB1 agonists. Therefore, it has been proposed that CB2 agonists are unlikely to be psychoactive or addictive. Thus, these may prove to be ideal candidates to provide analgesia independent of unwanted side effects.
There are some biological effects reported for cannabinoids that have been found to be independent of CB1 or CB2 receptor activity. These have since been attributed to GPR55 such as (-)-cannabidiol. GPR55 is also activated by the cannabinoid ligand CP 55,940 and a cannabidiol analog O-1602. The GPR55 receptor antagonist O-1918 blocks nociceptive firing in afferent C-fibers, and this suggests that manipulating GPR55 is a valid pain therapeutic target.
Tocrifluor T1117 is a useful tool developed to label GPR55 receptors in vitro fluorescently. Tocrifluor T1117 is a 5-TAMRA fluorescently labeled form of AM 251, that fluoresces at 543 nm excitation and can thus be used to help identify the neuronal expression of the GPR55 receptor (Figure 2).
Figure 2. Fluorescent ligand binding in the perivascular nerve. The fluorescent ligand, Tocrifluor T1117 (Cat. No. 2540), shows GPR55 receptor (red/orange) expression in perivascular nerve cells and blood vessels, additional staining illustrates expression of α1‑adrenoceptors (green). Image kindly provided by Dr. Craig Daly, University of Glasgow (see Daly et al (2010), for further reference).
Cannabinoid Modulation
One of the latest approaches is to target endogenous cannabinoids and their tonic influence over nociception in the CNS. This is in addition to treating pain states by targeting cannabinoid receptors. A transporter that has yet to be identified (endogenous cannabinoid transporter – ECT) reportedly facilitates reuptake of endocannabinoids from the synaptic space, whereby they are broken down within the cell.
However, AM 404, OMDM-2, UCM 707, and VDM 11 are inhibitors of this unidentified transporter that have been developed pharmacologically. In rodent pain models, these have demonstrated antinociceptive effects in neuropathic and inflammatory pain. Thus, a novel mechanism for alleviating symptoms of pain is through the systemic administration of these putative transport inhibitors – which increases levels of naturally occurring endocannabinoids AEA and 2-AG in the brain.
Endocannabinoids are degraded by enzymes such as fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL) within the cell. As shown in Figure 1, FAAH hydrolyzes AEA to arachidonic acid (AA), whereas 2-AG is metabolized to AA by MAGL. FAAH inhibitors such as MAFP, PF 750, arachidonyl serotonin and JNJ 1661010, have historically been known to possess antinociceptive properties, and are thus effective in reducing allodynia and hyperalgesia.
MAGL inhibitors, such as JZL 184 and N-arachidonyl maleimide, can elevate levels of endocannabinoids in the body and therefore can further provide a beneficial effect in boosting natural pain relief mechanisms. In fact, the inhibition of DAGL, which is an enzyme that catalyzes the conversion of 1,2-diacylglycerol (DAG) into 2-AG using the inhibitor, RHC 80267 can also prove to be useful for examining the role of endogenous cannabinoids.
In the CNS, the proposed tonic influence of endocannabinoids could potentially be targeted by the inactivation of FAAH, which is the primary enzyme responsible for cannabinoid breakdown. Such a scenario has been demonstrated in FAAH knockout mice that have reduced hyperalgesia. Meanwhile, though endocannabinoids seemingly exert a moderate influence on pain pathways, elevating their endogenous levels by inhibition of FAAH or manipulating endocannabinoid transporters can provide a therapeutic strategy for modulating endocannabinoid tone. Such a move could have significant effects on analgesia in pain models, minus the side effects associated with CB1 agonists, thus making endocannabinoids a potential target for modulating long term pain.
Opioids
One of the well-established classical treatments for pain are opioids, which remain one of the most effective targets in pain therapeutics to the present day. Opioid receptors are found both within the CNS, as well as throughout peripheral tissues. Opioids are G-protein-coupled receptors that are stimulated by endogenous peptides – such as endorphins, enkephalins, and dynorphins, and are currently classified into four categories: kappa (κ), delta (δ), mu (µ) and NOP receptors, with each group having multiple subtypes.
Opioid, as a term, represents any substance that produces morphine-like effects and can be blocked by antagonists such as naloxone. Although opioids possess almost unmatched effectiveness in easing pain, they suffer from serious side effects, which include nausea, constipation, respiratory depression, sleepiness, depression, hallucinations, and dependence, owing to their highly addictive properties.
By binding to inhibitory G-proteins (Gi/o), opioids inhibit adenylyl cyclase, reducing intracellular cAMP levels. They also display analgesic properties through direct coupling to ion channels, specifically by opening potassium channels and inhibiting calcium channel opening. Overall, this produces an effect of reduced neuronal excitability and a decrease in the release of pain neurotransmitters, resulting in analgesia.
When in pure form, opioid agonists (e.g. morphine, hydromorphone, and fentanyl) stimulate µ receptors and are thus the most potent analgesics. Contrastingly, partial agonists such as buprenorphine or pentazocine exhibit a ‘ceiling effect’. In other words, they have no further effect on pain beyond a particular dosage.
The state of analgesia can also be achieved through the targeting of specific subtypes of opioid receptors. In rodents, for instance, the highly selective δ opioid receptor agonist SNC 80 produces both antinociceptive and antidepressant effects, whereas endomorphin-1 and endomorphin-2 behave as potent analgesic peptides displaying the highest affinity and selectivity for the µ opioid receptor. Also, the µ opioid agonist DAMGO produces long-lasting antinociceptive effects in a rat model of neuropathic pain. Finally, NOP novel opioid receptor-like 1 (ORL1) receptor antagonists JTC 801 and (±)-J 113397, are capable of exhibiting potent antinociceptive effects in animal models of acute pain.
Glutamate (Metabotropic) Receptors
mGluRs or Glutamate metabotropic receptors are also treated as therapeutic targets in pain. These are G-protein-coupled receptors which are primarily split into three groups: mGlu Group I-III. The glutamate binding to the extracellular region of anmgluR is responsible for activating G-proteins bound to the intracellular region of the receptor to be phosphorylated. In turn, this then affects multiple intracellular pathways in the cell (Figure 1).
Further, compounds that can selectively target the mGluR subtypes have shown an ability to block pain pathways. These include the selective mGlu1a receptor antagonist, LY 367385 and highly potent group II antagonist LY 341495. Evidence also suggests that mGluR agonists, such as the mGlu2 receptor agonist LY 379268 andmGlu3 agonist L-AP4, can reverse pain pathways that are locked in a sensitized state, thus demonstrating a novel approach to targeting persistent pain.
Therefore, investigators can now begin to determine the therapeutic potential of the glutamate receptor in persistent pain states along with the ionotropic glutamate receptors owing to advances in the identification of compounds that are subtype-selective and systemically active.
GABA
As seen in Figure 1, the GABAB receptor is a metabotropic receptor that is linked to potassium channels via G-proteins. The alteration of the potassium concentration in the cell leads to hyperpolarization, which prevents sodium channels from opening and action potentials from firing. Thus, GABA is considered inhibitory receptors.
The GABAB receptor agonist (RS)-Baclofen has been seen to display analgesic properties, while the GABAB antagonist CGP 55845 blocks the antinociceptive actions of cholinergic agents.
In a rat model of sciatic nerve injury, the inhibition of GABA uptake by the GABA transporter GAT-1 is a target as well with NNC 711, a GAT-1 inhibitor, displaying an ability to induce analgesia. As suggested by this evidence, stimulation of GABA receptors at certain sites to offset the sedative side effects can prove to be beneficial in pain management.
Adenosine and P2Y Receptors
A G-protein-coupled receptor is the adenosine receptor (also categorized as a purinergic P1 receptor) which has recently been associated with pain research. Several adenosine receptor agonists have progressed through clinical trials, highlighting the fact that adenosine is a potential target for new drug development in pain. There are four main types of adenosine receptors: A1, A2A, A2B, and A3. All of these couple to G-proteins.
In functional studies, peripherally administering A1 and A2A receptor agonists such as N6-Cyclopentyladenosine and CGS 21680 respectively, leads to antinociception against hyperalgesia. A2B and A3 receptors primarily play a role that seems to be largely associated with inflammatory pain. Recently, it has also become evident that the metabotropic P2Y receptor can be found on sensory afferents and thus can have a role in modulating pain transmission.
Furthermore, P2Y receptors are G-protein-coupled receptors that are in the same family as the ionotropic P2X receptor. As revealed by in vivo studies, the administration of the P2Y2/4 agonist UTPγS inhibits pain transmission. Further, it has also been observed that the P2Y1 agonist MRS 2500 reduces hyperalgesia, suggesting that the P2Y receptor could also potentially become an important pain target.
Substance P and Tachykinin Receptors
Both the neuropeptide substance P and its closely related neuropeptide, neurokinin A, are believed to play an important physiological role in nociception modulation. These two are involved in relaying the intensity of noxious or painful stimuli. However, these can also be released from the peripheral terminals of sensory nerve fibers in the skin, muscle, and joints to initiate pain.
Further, substance P is a ligand for the tachykinin seven-transmembrane GPCR family, and its endogenous receptors are called neurokinin 1 (NK1) and NK2. In primary afferents, substance P coexists with glutamate that responds to painful stimuli. Finally, the administration of NK1 antagonists such as L-732,138 and RP 67580 has also been shown to attenuate hyperalgesia in rats, although this has not been proven clinically.
Bradykinin Receptors
Although it is considered as a peripherally acting inflammatory mediator, Bradykinin has also been shown to play a vital role in pain transmission. Released from kininogen precursors at the site of tissue injury and inflammation, Bradykinin acts on G-protein-coupled bradykinin receptor subtypes in primary sensory neurons. While the bradykinin 1 (B1) receptor is activated by injury, the B2 receptor is constitutively active.
B2 agonists including bradykinin have been shown to induce a state of thermal hyperalgesia, while B2 antagonists such as HOE 140 are known to reduce pain sensation in the formalin test of pain hypersensitivity. The B1 receptor, which is usually absent from non-inflamed tissue and with a low affinity for bradykinin, plays a lesser role in pain transduction. However, as demonstrated with B2 agonists, B1 receptor agonists such as Lys-[Des-Arg9] Bradykinin also produce a nociceptive response. Meanwhile, B1 receptor antagonists such as R 715 can prove be useful for investigating further the role of the B1 receptor in nociception.
About Tocris Bioscience
Tocris Bioscience is your trusted supplier of high-performance life science reagents, including receptor agonists & antagonists, enzyme inhibitors, ion channel modulators, fluorescent probes & dyes, and compound libraries. Our catalog consists of over 4,500 research tools, covering over 400 protein targets enabling you to investigate and modulate the activity of numerous signaling pathways and physiological processes.
We have been working with scientists for over 30 years to provide the life science community with research standards, as well as novel and innovative research tools. We understand the need for researchers to trust their research reagents, which is why we are committed to supplying our customers with the highest quality products available, so you can publish with confidence.
Tocris is part of the protein sciences division of Bio-Techne, which also includes the best in class brands R&D Systems, Novus Biologicals, ProteinSimple, and Advanced Cell Diagnostics. Bio-Techne has united these brands to provide researchers with a full portfolio of research reagents, assays, and protein platforms. For more information on Bio-Techne and its brands, please visit bio-techne.com.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices, and treatments.