Cellular signaling pathways control the proliferation, differentiation, survival, and migration of normal cells. But, dysregulation of these cells can cause tumors to form and grow. The overexpression or mutation of cell surface receptors and intracellular enzymes, along with the altered expression of growth factors, cytokines, and steroid hormones can promote the development and growth of malignancies, and trigger parenchymal cells to aid the development of tumors.
Moreover, mutations of components within intracellular signaling pathways can cause activated pathways that are unresponsive to external signals working to inhibit proliferation and apoptosis.
Growth Factor Receptors
Under normal physiological conditions, growth factor availability normalizes the balance between cell creation and division and cell death. This role in cellular homeostasis is often destabilized in cancer cases. Tumor cells can develop the ability to produce and secrete growth factors to stay alive, as well as make use of non-malignant cells to aid growth and avoid being identified by the immune system.
Growth factors bind transmembrane receptors, which contain inherent kinase activity. When activated, growth factors stimulate intracellular signaling pathways, including phosphoinositide 3-kinases (PI 3-K) and mitogen-activated protein kinase (MAPK) pathways (Figure 1).
In several human cancers, receptor tyrosine kinases (RTKs) are often affected by mutations causing upregulation of their signaling output (Figure 1). For example, the amplification or overexpression of the HER2/Neu/ERBB2 gene is recurrently seen in breast cancer cases (Figure 2).
HER2 is part of the ErbB family of RTKs, made up of four members: epidermal growth factor receptor (EGFR or ErbB1), HER2 (ErbB2), ErbB3 and ErbB4. Intracellular signaling from ErbB homo- and heterodimers occurs through the PI 3-K and MAPK signaling pathways (Figure 1).
Compounds that specifically target EGFR or ErbB2 such as iressa or TAK 165 respectively, are both clinically relevant and important tool compounds for the investigation into ErbB family signaling.
HKI 357 is a powerful inhibitor of both ErbB2 (HER2) and EGFR, which suppresses ligand-induced EGFR autophosphorylation and cell growth and division. This compound has also been proved to evade resistance to iressa in lung cancer cells. AG 490 also inhibits both EGFR and ErbB2 as well as JAK2, JAK3/STAT, JAK3/AP-1, and JAK3/MAPK pathways, demonstrating powerful inhibition of cytokine-independent cell growth in vitro and tumor cell invasion in vivo. Additionally, this compound selectively brings on apoptosis of leukemia cells over normal hematopoietic cells.
Type I insulin-like growth factor receptor (IGF1R) is significant in the development and endurance of several cell types and is universally expressed throughout the body. Dysregulation of IGF1R expression and signaling has been proved to drive tumor formation and progression. For example, studies have revealed that overexpression of IGF1R promotes the growth of tumors in mouse models and inhibits apoptosis in prostate cancer cells.
Key IGF1R inhibitors include BMS 536924, a dual inhibitor of the insulin receptor (IR) and IGF1R, which has been shown to inhibit receptor autophosphorylation and downstream MEK1/2 and Akt signaling.
BMS 536924 induces G1 arrest and cell death in acute myeloblastic leukemia (AML) cells and also prevents the proliferation of cells in several other tumor types. This compound also suppresses Snail mRNA expression in breast cancer cells that are overexpressing IGF1R; this results in increased levels of E-cadherin and the reversal of epithelial to mesenchymal transition (EMT) (Figure 1). GSK 1838705 is another IR and IGF1R inhibitor capable of blocking the creation and division of cancer cell lines in vitro and causes complete regression of ALK-dependent tumors in vivo.
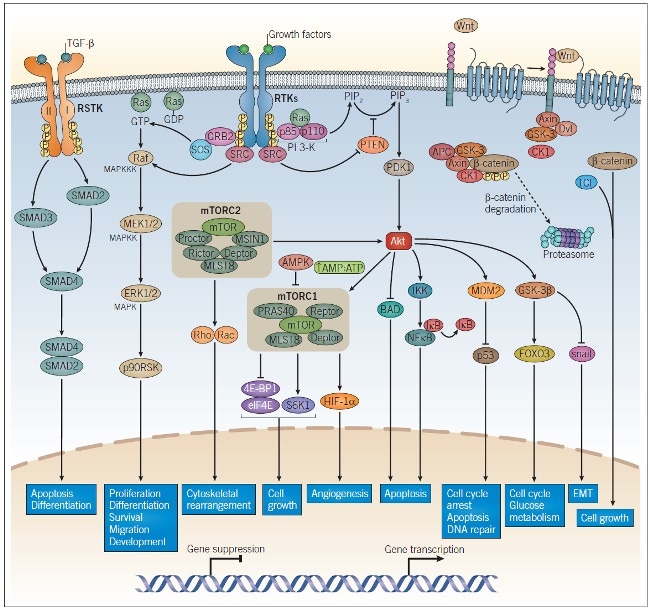
Figure 1: Intracellular Signaling Pathways in Tumorigenesis and Cancer Progression.
RTK growth factor receptors, TGF-β receptors, and Wnt signaling all play a role in tumorigenesis and cancer progression. Dimerization of growth factor receptors and TGF-β receptors occur upon ligand binding, enabling activation of the intracellular kinases on each receptor, leading to autophosphorylation. The phosphorylated residues on the cytoplasmic domain of the RTK bind adaptor proteins to initiate PI 3-K signaling and MAPK signaling. TGF-β binds the TGF-β receptor I/II dimeric complex and causes the activation of SMAD2 and SMAD3, which results in the association of SMAD2/SMAD4 and upregulation of TGF-β-responsive gene transcription. β-catenin undergoes proteasomal degradation in the absence of Wnt; upon Wnt binding, β-catenin is released from its complex and combines with TCF to promote Wnt-responsive gene transcription. These pathways regulate a wide range of cellular processes and encompass many of the major targets studied in cancer research. Abbreviations: RSTK: receptor serine-threonine kinase, RTK: receptor tyrosine kinases.
Anaplastic lymphoma kinase (ALK) is an RTK first identified in anaplastic large cell lymphoma (ALCL) as part of the fusion protein NPM-ALK. It has been shown that downstream effectors of ALK tyrosine kinase activity include the Ras-ERK, PI 3-K-Akt, JAK-STAT, and NF-κB signaling pathways.
In the absence of ligand binding, several ALK is inactive, and its expression promotes apoptosis. In contrast, when ALK is activated through either ligand binding or as part of an ALK fusion protein, rates of cell death is decreased. ALK promotes the development of tumors through overexpression and mutations leading to the development of certain functions.
ALK is overexpressed in lung cancer, melanoma, and certain types of breast cancer, among others, while point mutations in the ALK kinase domain have been linked to the development of neuroblastoma.
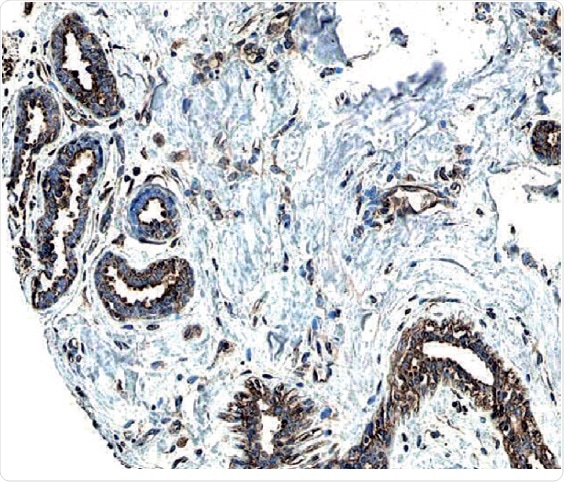
Figure 2: ErbB2/Her2 in Human Breast Cancer Tissue.
ErbB2 expression detected in paraffin-embedded sections of human breast cancer tissue. The ERBB2/HER2 gene is commonly amplified or overexpressed in breast cancer. The receptor is visualized here as brown staining using a Rabbit Anti-Human Phospho-ErbB2 Affinity-purified Polyclonal Antibody (R&D Systems, Catalog #AF4438). Hematoxylin counterstain in blue.
Inhibition of ALK using small molecules has shown promising preclinical results in lung cancer models. The powerful and orally active ALK inhibitors ASP 3026 and KRCA 0008 have been proved to reduce tumor growth in hEML4-ALK transgenic mice, and reduce the growth of xenograft lung cancer tumors in mice respectively.
Crizotinib is a potent dual inhibitor of ALK and mesenchymal-epithelial transition factor (c-MET), which displays antitumor efficacy in several tumor models, while also inhibiting c-MET-dependent proliferation, migration, and invasion of human tumor cells in vitro.
The high affinity and selective ALK and ROS1 inhibitor PF 06463922 have been shown to prevent the proliferation of a non-small lung cancer cell line containing a crizotinib-resistant ROS1 mutation in vitro. This compound is also orally available and brain penetrant and has been proved to inhibit tumor growth in relevant mouse models.
Fibroblast growth factor (FGF) signaling is mediated by FGF receptors (FGFR). This signaling pathway is regulated by FGFR expression levels and the affinity of FGF for the different FGFR isoforms.
In normal cells, FGF is a regulator in tissue homeostasis, tissue repair, and angiogenesis. Dysregulation of this pathway has been implicated in the development of tumors. Two useful compounds for studying the role of FGFR in cancer are PD 173074 and FIIN 1. PD 173074 is a selective FGFR1 and FGFR3 inhibitor, which has been shown to inhibit FGF and VEGF-induced blood vessel formation (angiogenesis) in a mouse cornea model of the process and to prevent tumor growth in lung cancer xenograft models. Derived from PD 173074, the potent, irreversible FGFR inhibitor FIIN 1 exhibit antiproliferative activity in FGFR3- and FGFR1-transformed Ba/F3 cells.
TGF-β is a growth factor that plays a key role in multiple pathways involved with cell adhesion, differentiation, motility, and death. In normal cells, TGF-β binds the serine/threonine kinase TGF-β receptors and makes cells less able to move through the cell cycle, as along with promoting cell death. Disruption of the TGF-β/SMAD pathway has been identified in a huge number of human cancers (Figure 1).
TGF-β is therapeutically beneficial in the early stages of cancer due to its ability to inhibit cell growth. However, in the more advanced stages of cancer, cells become refractory to TGF-β-mediated growth inhibition and instead enables the progression of tumor growth. In vitro studies have suggested that TGF-β enhances the invasiveness of cells by upregulating proteases such as MMPs and downregulating protease inhibitors.
As a tumor grows, the environment becomes hypoxic as a tumor grows and inflammation ensues, increasing TGF-β secretion by macrophages. This has been linked to metastasis. Moreover, TGF-β is involved in the induction of angiopoietin in premetastatic breast cancer cells, which encourages these cancer cells to remain in other tissues, such as the lungs.
TGF-β receptor kinase inhibitors have shown promising preclinical results. The strong and selective TGF-β receptor kinase inhibitor SB 431542, can inhibit TGF-β-induced EMT, migration, invasion, and VEGF secretion in several human cancer cell lines, as well as showing antitumor activity in a mouse model of colon cancer. A second strong compound is the TGF-β type I receptor inhibitor A 83-01. This compound blocks phosphorylation of Smad2 and inhibits TGF-β-induced EMT.
While some clinically relevant inhibitors are selective for individual RTKs or RTK families, others have proved effective by targeting multiple receptors. For example, sunitinib targets, among others, PDGFRb, VEGFR, FLT3, and RET. Other compounds, such as lestaurtinib, are other broad-spectrum inhibitors that target both the receptor and intracellular kinases. By combining EGFR inhibitors with inhibitors for other clinically relevant receptors such as IGF1R, TGF-β, or c-MET, it may be possible to overcome the resistance to selectively targeted agents that characterize certain types of cancers. Looking to the future, personalized or precision medicine will focus on developing tailor-made combinations of relevant drugs, depending on the individual genetic makeup of the patient.
About Tocris Bioscience
Tocris Bioscience is your trusted supplier of high-performance life science reagents, including receptor agonists & antagonists, enzyme inhibitors, ion channel modulators, fluorescent probes & dyes, and compound libraries. Our catalog consists of over 4,500 research tools, covering over 400 protein targets enabling you to investigate and modulate the activity of numerous signaling pathways and physiological processes.
We have been working with scientists for over 30 years to provide the life science community with research standards, as well as novel and innovative research tools. We understand the need for researchers to trust their research reagents, which is why we are committed to supplying our customers with the highest quality products available, so you can publish with confidence.
Tocris is part of the protein sciences division of Bio-Techne, which also includes the best in class brands R&D Systems, Novus Biologicals, ProteinSimple, and Advanced Cell Diagnostics. Bio-Techne has united these brands to provide researchers with a full portfolio of research reagents, assays, and protein platforms. For more information on Bio-Techne and its brands, please visit bio-techne.com.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices, and treatments.