The prototype of the cys-loop family of ligand-gated ion channels is nicotinic acetylcholine receptor (otherwise known as nAChR). This family also includes GABAA-ρ {formerly GABAC}, glycine and 5-HT3 receptors, in addition to invertebrate glutamate-, histamine-, ACh-, and 5-HT-gated chloride channels.1–3 Recently, we have seen the identification of homologous ligand-gated ion channels (despite their lack of the hitherto definitive ‘cys-loop’) in prokaryotes, and it is likely that they represent the ancestral form preceding current day ‘cys-loop’ receptors.4,5
nAChRs constitute a protein family which serve myriad physiological functions:
- postsynaptic nAChRs mediate skeletal muscle contraction at the neuromuscular junction;
- ganglionic nAChRs are responsible for fast excitatory transmission in the autonomic nervous system; as well as being located in presynaptic nerve endings of sympathetic, parasympathetic and sensory neurons;
- throughout the brain and spinal cord, ‘neuronal’ nAChRs are present and exert a mostly modulatory influence;
- on non-neuronal cells, ‘neuronal’ nAChRs including glial immune and endothelial cells also occur, where they are presumed to respond to paracrine ACh.
Within this receptor family, the structural and functional diversity6 has sparked interest in nAChRs as potential therapeutic targets to address various medical conditions, and has spurred drug discovery programs. The result of this has been the development of subtype-selective ligands that complement the generous natural product resources directed at nAChRs, in order to facilitate an increasing portfolio of tools for research into nAChRs.7,8
nAChR Structure
The first receptors to be studied and characterized in exquisite functional and structural detail were nAChRs in vertebrate skeletal muscle, and their counterparts in the electrogenic organs of Torpedo and Electrophorus. In early studies by Langley an Dale, which followed the pioneering work of Katz and Miledi as well as the development of single channel recording by Neher and Sakmann, the neuromuscular junction enabled detailed electrophysiological measurements of nAChR function to be made.1,8
A high density of nAChRs were provided by Torpedo and Electrophorus electric organs, and these facilitated high-resolution structural studies9,10 and biochemical characterization.1 Snake α-toxins with high affinity, notably α-bungarotoxin (α-Bgt), allowed the purification of the nAChR protein from Torpedo electroplax and resolution into 4 different subunits, designated α,β,γ and δ.11 Subsequently, an additional subunit, ε, was identified in nAChRs purified from adult skeletal muscle.
These subunits were cloned in the early 1980s, and the day of the molecular analysis of nAChR dawned.1 The muscle endplate nAChR has the subunit combination and stoichiometry (α1)2β1εδ, whereas those extrajunctional (α1)2β1γδ nAChRs are predominantly located in fetal or denervated muscle, and electric organs.12
Each of the five subunits which comprises the nAChR creates a water-filled pore by spanning the lipid bilayer (Figure 1). Each subunit is made up of 4 transmembrane segments; the second transmembrane segment (M2) lines the ion channel. Every subunit’s extracellular N-terminal domain possesses a ‘cys-loop’ , which signifies the characteristic sequence of this ligand-gated ion channel family: two cysteine residues (Cys 128, 142, Torpedo α1 subunit numbering), separated by 13 amino acids, generate a disulfide bond to form a loop implicated in the transduction of agonist binding into channel opening.13,14
The N-terminal domain of α subunits houses the principal agonist binding site, near the pair of adjacent (‘vicinal’) cysteine residues (Cys 192, 193, Torpedo numbering) that are characteristic of an α subunit. The importance of 4 aromatic residues has been highlighted by mutagenesis and photoaffinity labeling experiments (Tyr 93, Trp 149, Tyr 190, Tyr 198, Torpedo numbering), which are consistent with 3 polypeptide loops of the α subunit (loops A–C) and contribute to the principal agonist binding site.14,15 Through π-cation interactions, these aromatic residues stabilize bound ligands. A clockwise order arranges the muscle nAChR subunits– α1, β1,δ,α1,γ/ε – with the two agonist binding sites occurring at the α-δ and α-γ/ε interfaces12 (see Figure 3).
The agonist binding site is also contributed by the adjacent subunit (δ or γ/ε) (complementary site: polypeptide loops D-F). A result of this is that the α-δ and α-γ/ε binding sites are not identical, a fact which is reflected in differences in ligand affinity.12,22 However, for effective opening of the channel, occupancy of both binding sites is required. The knowledge of ligand binding to nAChRs has been hugely ameliorated by the identification of a crystal structure of a soluble ACh binding protein (AChBP). This structure was identified first in the snail Lymaea stagnalis and subsequently cloned from Aplysia californica and Bulinus truncatus. 16–18
Every subunit of this pentameric secreted protein is similar to the N-terminal domain of a nAChR subunit (20–24% sequence identity), with conservation of all the residues implicated in ACh binding to the muscle nAChR. The capacity to crystallize these proteins for X-ray diffraction studies has offered a high-resolution imaging of the extracellular portion of the receptor, significantly of the binding site at the interface between adjacent subunits, and the interaction between these sites and ligands.14,17,19
Both co-crystallization of the AChBP with different ligands, as well as molecular modeling approaches which modify the crystal structure of the AChBP (to accommodate sequence differences found in the extracellular domains of various nAChR subunits) can be used to identify subtype-selective nAChR ligands.20,21
Where the function of the AChBP is to sequester ACh, binding of ACh to nAChRs increases the likelihood of channel opening, which promotes cation flux. At the moment of agonist binding, nAChRs encounter an allosteric transition from the largely closed, resting conformation to an open state that allows an influx of Na+, and, to a certain degree (depending on nAChR subtype, see Figure 3), Ca2+, accompanied by an efflux of K+ under typical physiological conditions.
The ion channel, in the closed state, is occluded by a ‘hydrophobic girdle’ which creates a barrier to ion permeation.9 In the extracellular domain, agonist binding promotes a conformational change resulting in a rotational movement of the M2 helices lining the pore, rotating the girdle to broaden the pore by ~3 Å, suitable for ion permeation.9,23 The ensuing depolarization, at the muscle endplate, elicits muscle contraction. Although agonist is present, the nAChR channel closes from seconds to minutes, to enter a desensitized state (Figure 2). The nAChR is refractory to activation in this condition. It has been proposed that myriad desensitized states exist.24
The nAChR binds agonists with low affinity (e.g. Kd of muscle nAChR for ACh ~50 μM) in the open and active conformation. Higher affinity is displayed for agonist binding by the desensitized states (Kd of muscle nAChR for ACh ~1-5 μM); therefore, the desensitized nAChR retains bound agonist in spite of its non-conducting state. Allosteric modulators can influence the transitions between resting, open and desensitized states (see below).
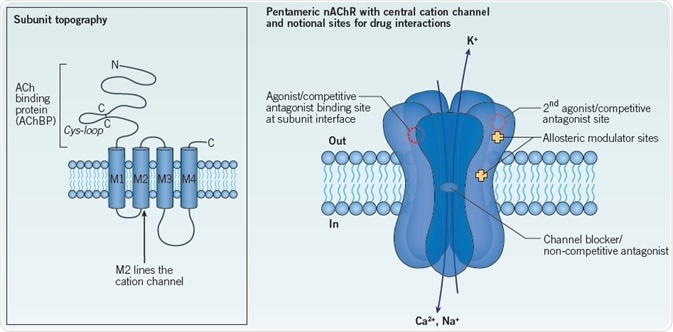
Figure 1. General structure of a nAChR constitute a principal agonist binding site in neuronal nAChRs.
Left: The topography of a single nAChR subunit. The two cysteine residues forming the signature cys-loop are indicated. The extracellular N-terminal domain corresponds to the AChBP.
Right: Schematic of a generic nAChR showing 5 subunits spanning the membrane to form a central ion channel. Notional sites of action of interacting drugs are shown: two agonist binding sites are indicated at extracellular subunit interfaces, consistent with a heteromeric nAChR. Competitive antagonist sites overlap the agonist binding sites. Extracellular and intramembrane sites have been proposed for different allosteric modulators. Non-competitive antagonists may occlude or block the ion channel, as indicated, or may act at other sites. See text for further details. Image credit: Tocris Bioscience
Sites on the nAChR for Ligand Interactions (Figure 1)
As detailed above, agonists bind at subunit interfaces in the extracellular domain.14 These agonists stabilize the mobile loop-C of the principal binding subunit in close apposition to the adjacent (complementary) subunit. In addition, competitive antagonists also bind either at or near to the agonist binding sites, which blocks access to agonists. Contrastingly to agonists, competitive antagonists stabilize loop-C in an extended conformation that prevents the channel from opening.17,19,25 Therefore, the fact of the channel remaining closed ensures that access to the agonist binding site is prevented.
Unlike full agonists, and despite interacting with the same agonist binding site, partial agonists activate the nAChR with low efficacy, as exemplified by the endogenous ligand ACh.26 Ligands like this additionally display competitive inhibition through competition for occupancy of the binding site with other, more efficient agonists.
Therefore, as evidenced, they combine a degree of antagonism with weak agonism.8 Though this efficacy displayed by a partial agonist is not the ligand’s intrinsic property, it is instead a consequence of the interaction with a particular nAChR subtype.139 For instance, the efficacies of synthetic agonists TC 2559 and sazetidine A at α4β2 nAChRs are determined by the subunit stoichiometry.27 The major determinant of efficacy has been indicated to be the complementary subunit, based on a comparison of cytisine and a novel agonist NS 3861.37
To prevent receptor activation or to block channel function, non-competitive antagonists bind at sites distinct from agonist binding sites. Compounds which block channels may interact specifically with lumen of the pore or residues in the mouth in order to occlude the ion channel. Additionally, we may predict that any small, positively charged species channel block, and that many agonists, including ACh, do this at high concentrations.28 As access to the channel requires the channel to be open, the efficiency of channel blockade is ‘state dependent’. Therefore, the state of the receptor (resting, open, desensitized) will influence the speed of block (Figure 2).
Notional sites of action of interacting drugs are shown: two agonist binding sites are indicated at extracellular subunit interfaces, consistent with a heteromeric nAChR. Competitive antagonist sites overlap the agonist binding sites. Extracellular and intramembrane sites have been proposed for different allosteric modulators. Non-competitive antagonists may occlude or block the ion channel, as indicated, or may act at other sites. See text for further details.
Allosteric modulators are the names given to a series of non-competitive ligands which act at a myriad of separate sites in order to influence, either positively or negatively, agonist interactions and/or nAChR function.29 This class includes such diverse members as inorganic cations, steroids, anesthetics and amines. In particular, subtype-selective positive allosteric modulators (PAMs) are garnering attention as potential therapeutic drugs.8 Their sites of interaction on the nAChR are diverse, which span from non-canonical binding locations in the extracellular N-terminus to locations within the membrane spanning domains (Figure 1)30–32
Diversity of nAChR Subtypes (Figure 3)
Paton and Zaimis’s classical studies 33 displayed the fact that nicotinic responses in autonomic neurons are pharmacologically distinct from their skeletal muscle counterparts, which offers the first indication of nAChR heterogeneity. The quest to explain the psychoactive actions of nicotine, and subsequently for nAChRs in the brain, has identified binding sites for 125I-α-Bgt and [3H]-nicotine.34,35
The (then) novel and controversial concept of nAChR heterogeneity was raised by their distinct pharmacological profiles and anatomical distributions in rodent brain. Eleven novel ‘neuronal’ nAChR subunits have been identified in mammals (α2- α7, α9, α10, β2-β4), with an additional subunit, α8, cloned from avian species, after the first publication36 of a cloned neuronal nAChR subunit (α3) in 1986.6,8
α2-α6 subunits create heteromeric nAChRs with β subunits. The characterization of α subunits is the presence of a pair of vicinal cysteines, which equate to those that characterize the muscle α1 subunit. This gave rise to the suggestion that all α subunits could constitute a principal agonist binding site in neuronal nAChRs. However, this is not the case: the α5 subunit cannot fulfil this role due to its lack of critical tyrosine from loop C (Tyr 190, Torpedo α1 labeling).38 β subunits do not have the N-terminal vicinal cysteines; however, β2 and β4 subunits possess the tryptophan residue characteristic of loop D; therefore, these subunits can act like γ and δ muscle nAChR subunits to offer the complementary agonist binding site at an αβ interface.
The lack of this key tryptophan residue in the β3 subunit makes it the true homolog of the muscle β1 subunit that does not contribute to an agonist binding site. Certainly, the similarity in sequences between α5 and β3 subunits468 (see Figure 3) corroborates the idea that both have this role, though other subunits also have the capability to occupy the position equivalent to β1.39,40 Hurst et al. review issues of nAChR subunit nomenclature. 8
nAChR Stoichiometry
Heteromeric neuronal nAChRs made up of α and β subunits incorporate two pairwise combinations of α2, α3, α4 or α6 with β2 or β4 subunits, to offer two agonist binding sites in an arrangement which is analogous to muscle nAChR (Figure 3).
The fifth position, corresponding to that occupied by the β1 subunit in muscle nAChR, can accommodate any subunit (though rules of assembly restrict the subunit combinations in a given nAChR, Figure 3). The subunit which occupies this position (also known as the ‘accessory’ subunit)40 has the power to influence assembly, trafficking and functional properties of the resultant nAChR subtype. This includes agonist potency, conductance, Ca2+ permeability and desensitization kinetics.27,40
For instance, agonists and competitive antagonists generally boast a higher potency at (α4)2(β2)3 nAChRs, in comparison with (α4)3(β2)2 nAChRs.27 Nicotine upregulates α4β2 nAChRs through acting as a chaperone to stabilize the (α4)2(β2)3 stoichiometry.41,42 α3β4 nAChRs additionally display stoichiometry-dependent divergences in agonist and antagonist sensitivities and nicotine-induced upregulation, in heterologous expression systems.37,43,44
The fifth subunit contributes to a putative allosteric site, which is comparable to the GABAA receptor benzodiazepine binding site.14 Some evidence exists to corroborate the concept that allosteric modulators bind to neuronal nAChRs here, depending on which subunit occupies the fifth position. For instance, at this site, the anthelmintic morantel acts in mammalian α3β2 nAChRs to enhance channel gating.30
Site-directed mutagenesis, combined with cysteine substitution methods, imply that β2 occupies the fifth position and thereby creates non-canonical binding site (β2 – principal face; α3 – complementary face).30 In contradiction to this, it is proposed that the PAM NS 9283 interacts selectively with an α4β2 heteromer with stoichiometry (α4)3(β2)2, placing α4 in the fifth position.46 Nonetheless, there exists only a poor establishing of the precise subunit stoichiometry of native nAChRs.
Contrastingly, the α7, α8 and α9 subunits are characterized by their capacity to form robust homomeric receptors which are sensitive to α-Bgt, when expressed in Xenopus oocytes. The evidence indicates that in mammalian brain, native α-Bgt-sensitive nAChRs are predominantly homomeric, and are formed of α7 subunits.6 In comparison with heteromeric nAChRs, despite this, heterologous expression of α7 nAChRs is much less efficient in non-neuronal mammalian cultured cells.
The resolution of this paradox arose thanks to the discovery that the endoplasmic reticulum-resident chaperone RIC-3 can promote the formation of functional α7 nAChRs in nonpermissive cells.47 This underlines the significance of ancillary proteins in the regulation of the assembly, trafficking, stabilization and turnover of nAChR subtypes.48 The same type of subunit provides both principal and complementary faces of the agonist binding site in a homopentamer,66 which results in five putative binding sites per receptor homomer (Figure 3).
There exists an intriguing question regarding the proportion of sites which must be occupied by an agonist in order to obtain efficient channel activation. To determine the single channel activity of receptors composed of different proportions of wildtype and mutated (low conductance) subunits, studies demonstrated that for full channel activity, occupancy of 3 non-consecutive sites within a pentamer is necessary.49
Despite its capacity to form homomeric nAChRs, in physiological systems, despite its ability to form homomeric nAChRs the α9 subunit co-assembles with α10 to form heteromeric nAChRs, with the stoichiometry (α9)2(α10)3.50,51 When on its own, α10 seems to be unable to form a homomeric receptor.52 Heteromers exist in avian tissues α7α8, and it has been proposed that nAChRs made up of α7 and β2 subunits occur in heterologous expression systems and in mammalian brain.53–55
Invertebrate nAChRs
A gene family of α and β nAChR subunits has been uncovered in invertebrates, which are distinct but related. The largest number of such subunits is incorporated by the C. elegans genome sequence, with to date at least 32 reported, of which 22 have been classified as α subunits.56 This contrasts with a lower number of subunits in parasitic nematodes, wherein they are clinical and veterinary targets.56 A number of insects, like the honey bee (Apis mellifera) fruit fly (Drosophila melanogaster), and malaria mosquito (Anopheles gambiae), all have 10–12 nAChR subunits that make up the major excitatory receptor class within the insect central nervous system.57,58
The development of selective agonists has been allowed by this distinct gene family of nAChR subunits - the neonicotinoids that target insect or helminth nAChRs, for pesticide or veterinary applications.59 Electrophysiology, mutagenesis, and computer modeling indicate that the subunit supplying the complementary face of the binding site confers the selectivity for neonicotinoids of invertebrate over mammalian nAChRs. There are concerns regarding the potential impact of neonicotinoids on ‘friendly’ insects, like bees: these fears underline the significance of the task to generate more discriminating ligands that are able to determine between insect species.60
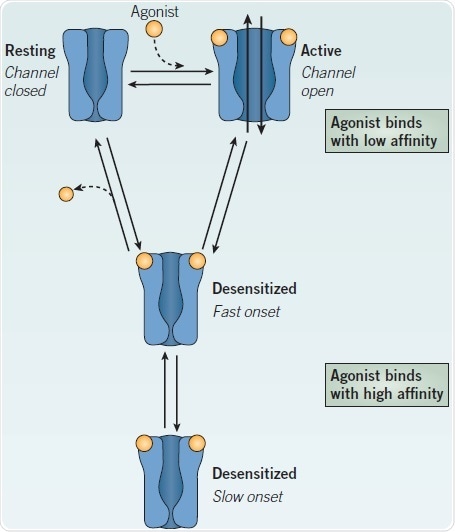
Figure 2. Relationship between the major conformational states of a nAChR. Image credit: Tocris Bioscience
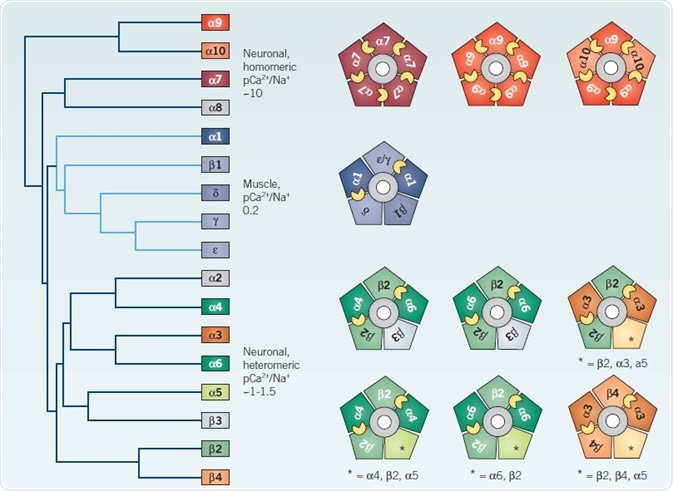
Figure 3. Heterogeneity of vertebrate nAChRs. Image credit: Tocris Bioscience
Left: Cladogram of all vertebrate nAChR subunits cloned to date, adapted from Novère et al 468 Right: Cartoons illustrate viable pentameric mammalian subunit combinations, with putative agonist binding sites indicated by small yellow crescents between adjacent subunits. See text for further details. *Indicates the possible inclusion of additional unspecified subunits
Physiological Significance and Distribution of nAChR Subtypes
Autonomic neurons typically express α3, α5, α7, β2 and β4 subunits,61,62 with the likely assembly of α3β4, α3β4α5, α3β4β2 and α7 nAChRs (including sympathetic ganglia, parasympathetic innervation, sensory ganglia, chromaffin, neuroblastoma and PC12 cells). Further additional subunits have been reported in dorsal root ganglia;63–65 nAChRs in these sensory neurons capture interest as potential therapeutic targets for modulating nociceptive signals (the subunits reported include α4, α6, α9 and α10). There is a heterogeneous distribution of α2-α7 and β2-β4 subunits within the mammalian brain, with clear and often extensive and overlapping expression patterns.8,66
The most widespread subunits are α4, β2 and α7, with α4β2* (where * indicates the possible inclusion of additional unspecified subunits),67 and α7 nAChRs possessing a somewhat complementary distribution. Contrastingly, to the clear and well-established role of nAChRs in synaptic signaling in both the neuromuscular junction and sympathetic ganglia, there is a scarcity of reports which detail neuronal nAChRs mediating cholinergic synaptic transmission in the CNS. In the brain, however, there is abundant evidence in the brain for presynaptic nAChRs which can modulate the release of many different neurotransmitters68. This has led to the proposition that most nAChRs are located presynaptically.
nAChRs additionally exist in perisynaptic and extrasynaptic locations on somatodendritic regions,69,70,355. The current view held is that, in response to non-synaptic (‘paracrine’) levels of ACh (or choline, in the case of α7 nAChRs) presynaptic and extrasynaptic nAChRs modulate short and longer term neuronal activity.71,72 Nonetheless, several examples have been documented of synaptic nAChRs mediating synaptic transmission at cholinergic synapses in the brain.73,74 In the hippocampus and cortex (including the prefrontal cortex (PFC)), the α7 nAChR is particularly prominent,75 wherein it is commonly associated with GABAergic interneurons and glutamatergic synapses.72,77
There exist multiple reports of the highly Ca2+ permeable α7 nAChRs contributing to synaptic plasticity and long term potentiation, most significantly in the hippocampus,71,72,76,78 as well as featuring in working memory in the PFC.355 α7 nAChRs have been linked to schizophrenia by genetic linkage studies and pharmacological evidence.79 Much evidence has stimulated the aim of targeting α7 nAChRs in order to treat attentional and cognitive deficits in multiple conditions such as ADHD, schizophrenia, and Alzheimer’s disease.8 In modulating synapting activity, α7 and α4β2 nAChRs also collaborate.69,80 α4β2 nAChRs with stoichiometry (α4)2(β2)3 have high affinity for nicotine (and account for >90% of [3H]-nicotine binding to brain tissues81).
Nicotine self-administration is eliminated by transgenic knockout of either of these subunits, whereas virally targeted re-expression of the β2 subunit in mesolimbic areas of β2 knockout mice reinstates this behavior. This indicates that there is a role for α4β2 nAChRs in nicotine addiction.82,83
A role has been uncovered for β2* nAChRs in the prelimbic area of the PFC in attention, through a similar strategy.84 α4β2 nAChRs are highly expressed in the thalamus.85 Their assumed role in thalamo-cortical circuitry provides a rationale for the ability of gain of function mutations in the channel-forming M2 domain, or adjoining M3 domain, of either the α4 or β2 subunit to give rise to autosomal dominant nocturnal frontal lobe epilepsy.86
An evolutionarily conserved gene cluster is formed by α3, α5 and β4 nAChR subunit genes, which is found on human chromosome 15. These are often co-expressed.8 α3 and β4 nAChR subunits have a more restricted distribution in the CNS, compared with α4 and β2, but the α5 subunit is expressed more widely, at low to moderate levels.87–89 Polymorphisms in these 3 genes are associated with increased vulnerability to tobacco addiction and/or risk of lung cancer, notably D398N within the coding region of the α5 subunit gene.90 Thanks to this association, studies were undertaken in mice which implicate α5-containing nAChRs in the medial habenular – interpeduncular nucleus (IPN) pathway in the negative regulation of the consumption of nicotine. These make such receptors a potential target for smoking cessation.91,92
α5 is associated with α4 and β2 subunits in 15-40% of α4β2* nAChRs, in cortex, thalamus, hippocampus and striatum.89 Identification has taken place of somatodendritic or terminal α4β2α5 nAChRs in dopamine neurons in the ventral tegmental area (VTA) and substantia nigra and in corticothalamic projection neurons.93–95 α5 confers increased Ca2+ permeability and enhanced rates of desensitization to heteromeric nAChRs,40 are able to change sensitivity to ACh, and makes α4β2 nAChRs resistant to nicotine-induced upregulation.89,96–98
Commonly, α6 and β3 subunits are expressed together and display a restricted distribution, which is mostly limited to catecholaminergic neurons and neurons of the visual pathway.6,99,100 These subunits contribute to nAChRs of complex subunit composition on dopaminergic terminals, including α6β2β3 and α4α6β2β3 nAChRs93 (Figure 3). It is inferred that the latter subtype has the highest nicotine sensitivity (EC50 = 0.23 µM) and sazetidine A.101,102 It is suggested that the β3 subunit influences the assembly, stability and/or targeting of α6-containing nAChRs103–105 and from studies of recombinant nAChR subunits, a dominant negative role has been inferred.106,107
In the rodent CNS, the α2 nAChR subunit has a restricted expression pattern, as it is largely limited to the IPN.108 In the primate brain, its distribution seems to be more extensive.109 Transgenic mice which have a targeted deletion of the α2 subunit gene display minimal change in phenotype, though certain nicotine-modulated behaviors like withdrawal and nicotine self-administration are potentiated.110 This data remains consistent with the IPN role in constraining addictive behavior, which we have alluded to above.91,92 It is also inferred that α2β4 nAChRs also participate in the motorneuron-Renshaw cell synapse of the mouse spinal cord, which is a rare model of nicotinic synaptic transmission in the CNS.111
Mechanosensory hair cells express α9 and α10 nAChR subunits which co-assemble in order to generate nAChRs that are predominantly heteromeric. α9α10 nAChRs display a nicotinic/ muscarinic pharmacology which is unusual in that it is mixed, although they are activated by ACh, nicotine and other agonists act as competitive antagonists at α9α10 nAChRs.52,124
α9α10 nAChRs are highly Ca2+ permeable and mediate efferent olivocochlear system effects on auditory processing.51,112 Though these subunits are not expressed in the brain, they have been reported, either separately or together, in a variety of non-neuronal cells and tissues, and also in sensory neurons where they are considered to be antinociceptive targets.113
Analysis of diverse non-neuronal cells has also revealed the expression of numerous ‘neuronal’ nAChR subunits. These include the following: macrophages, astrocytes, keratinocytes, muscle cells, lymphocytes, endothelial cells of the vascular system, intestinal epithelial cells and various lung cell-types.114 The presence of mRNAs which encode most nAChR subunits has been detected in such cells; however, the identity and functional significance of assembled nAChRs in non-neuronal cells is less clear. α7 nAChRs in non-neuronal cells arouse interest thanks to their high Ca2+ permeability as well as their capacity to engage Ca2+-dependent signaling cascades, but for both sides of the argument compelling evidence exists, as to whether there are adverse or beneficial influences.115,116
Nicotinic Ligands for Neuronal nAChR
Nature has developed a diverse array of toxins, both plant and animal, that target these receptors and their CNS-counterparts, thanks to the critical roles of muscle and ganglionic nAChRs in normal vertebrate physiology. The generation of synthetic ligands, in response to the perceived validity of neuronal nAChRs as therapeutic targets continues to augment the nicotinic pharmacopoeia. Consequently, the following listing is primarily directed at neuronal nAChRs. In particular, the α7 nAChR has attracted a large number of antagonists, selective agonists, and allosteric modulators.
For heteromeric nAChRs, there remains a scarcity of subtype-selective tools, particularly ligands that can discriminate between nAChR subtypes with subtle differences in subunit composition and stoichiometry. The lack of synthetic subtype-selective antagonists I most significant, which potentially demonstrates the lack of therapeutic potential associated with nAChR blockade. The α-conotoxins afford some compensation for this deficit. The generation of subtype-selective PAMs has been a notable development. Below is a brief discussion of the representatives of these various classes of ligand that are currently available from Tocris.
See references 7, 8 and 117 for more comprehensive accounts of some of the families of synthetic nicotinic ligands.
Agonists
nAChR agonists enjoy structural diversity, as they comprise natural products from various plant and animal species, and synthetic molecules from structure activity relationship programs or drug library screening .8,117 Non-selective agonists like nicotine typically bind with highest affinity (Ki ) to the (α4)2(β2)3 nAChR in equilibrium binding studies, with 2–3 orders of magnitude lower affinity at α7 nAChRs and intermediate affinity at α3* nAChRs.
The high affinity desensitized state of the nAChR (Figure 2) is reflected in binding, as the former is stabilized during the prolonged incubation period of such assays. Compared with their affinities for α7 nAChRs, agonists are ~10-fold more potent at α9α10 nAChRs.118 EC50 values are typically 1–3 orders of magnitude higher than Ki values, with respect to functional potency. This difference is greatest for α4β2 nAChRs, so that EC50 values between subtypes may hold more similarities to one another than initially suggested by their binding affinities.
This information has ignited the spark for subtype-selective nAChR agonists to exclusively activate a particular subtype in a mixed population. Typically, the functional potency of novel ligands is determined by Ca2+ flux assays of recombinant nAChR subunits expressed in Xenopus oocytes or mammalian cells or electrophysiological recording.119,120,121,272 In native systems, evaluation is made complex by the likely presence of a multiplicity of nAChRs of complex and often unknown subunit composition. A practical aspect springs from the analysis method of rapidly desensitizing currents, significantly those recorded from cells expressing α7 nAChRs.
As is traditionally the case, measurements of the peak height (amplitude) of electrophysiological responses to construct concentration response relationships nonetheless underestimate the potency of agonists activating α7 nAChRs. This is due to the fact that concentration response relationships based on net change analysis (area under the curve) provide more accurate (and more potent) EC50values.122 For more slowly desensitizing nAChRs, like, these two methods are in good agreement with respect to EC50 values and Hill coefficients, for more slowly desensitizing nAChRs, like α4β2 and α3β4.123
Antagonists
There are very few nAChR antagonists, as stated. Antagonists have also been employed to generate animal models of impaired nicotinic function, as reviewed by Roegge et al, in addition to their use as tools for defining nAChR responses and mechanisms, in vitro and in vivo.252
While some newer synthetic molecules are starting to arise from SAR (structure-activity relationship) programs, most have a long pedigree, many emanating from natural products. The need for nAChR subtype-selective antagonists are emphasized by increasing awareness of the subunit complexity of native nAChRs. Nonetheless, it is that very complexity, including differences in stoichiometry and subtle species differences, that adds the challenging aspect to the identification and rigorous characterization of subtype-selective ligands.
Competitive Antagonists
By definition, the inhibition achieved by competitive antagonists can be overcome by increasing the agonist concentration through interacting with the nAChR at, or close to, the agonist binding site. Therefore, this competitive antagonism is referred to as ‘surmountable’, shifting the concentration response relationship for the agonist to the right (e.g. (+)-tubocurarine block of ACh-evoked currents in Xenopus oocytes; 253 dihydro-β-erythroidine (DHβE) versus nicotine-evoked [3H]-dopamine release).186 In consequence, experimental conditions, significantly the agonist concentration, will influence the degree of functional blockade achieved by a given concentration of competitive antagonist.
Future Perspectives for nAChR Research
Since the previous version of this review, the past seven years have offered monumental insights into understanding of the molecular structure of nAChRs, including a new understanding of their subunit stoichiometry, assembly and trafficking, and an new perspective on their cellular functions and roles in physiological and pathological processes.
However, each new layer of knowledge raises yet further questions. In order to facilitate the answers, many new nicotinic ligands have been published, and a positive increase in the number of nAChR subtypeselective agonists and PAMs has been seen.8 It is likely that these synthetic successes will pavev the way for more novel nicotinic probes. As an example, RuBi-Nicotine469 (page 9) is a caged nicotine which will have applications for the rapid, localized activation of nAChRs.
The possibility exists too of monitoring nAChR behavior via detection mechanisms coupled to subunit proteins, like Förster resonance energy transfer (FRET)471 or optochemical technology.472
Despite the fact that, for quantifying nAChRs, the use of radioligands has declined in recent years, we have seen a growth in the development of novel radioligands, (based on subtype-selective nicotinic ligands) for in vivo analysis of nAChRs by positron emission tomography (PET) studies.473,474 For identifying nAChRs, antibodies remain problematic (in brain tissue at least) 475 but alternative approaches are offered by peptide toxins.
A valuable marker for labelling muscle and α7 nAChRs in cells and tissue has been fuorescently-labeled α-Bgt,476,477 and, with some success, fluorescent labeling is being extended to the α-conotoxins with some success.478
Additional approaches are offered by biotinylated α-Bgt, for example, detection by fluorophore-labeled anti-biotin antibodies (for signal amplification)479 or single-particle tracking of nAChRs in live cells using streptavidin quantum dots.70,479,480 Certainly, we have seen the incorporation of α-Bgt binding motifs into α3 subunits483 and other membrane proteins481, in order to take advantage of α-Bgt-linked detection agents. The future, with these technological advances and new chemical entities, holds exciting prospects for resolution in terms of nAChRs and explication of their roles in health and disease.
Special Mentions
This white paper was based on the original work of:
Susan Wonnacott: Susan Wonnacott is a Professor of Neuroscience in the Department of Biology and Biochemistry at the University of Bath. Her research focuses on understanding the roles of nicotinic acetylcholine receptors in the mammalian brain and the molecular and cellular events initiated by acute and chronic nicotinic receptor stimulation.
Department of Biology and Biochemistry, University of Bath, UK.
Email: [email protected]
References
- Changeux (2012) J.Biol.Chem. 287 40207.
- Jones and Sattelle (2006) Invert.Neurosci. 6 123.
- Wolstenholme (2012) J.Biol.Chem. 287 40232.
- Tasneem et al. (2005) Genome Biol. 6 R4.
- Duret et al. (2011) Proc.Natl.Acad.Sci.U.S.A. 108 12143.
- Millar and Gotti (2009) Neuropharmacology 56 237.
- Gündisch and Eibl (2011) Expert Opin.Ther.Pat. 21 1867.
- Hurst et al. (2013) Pharmacol.Ther. 137 22.
- Unwin (2013) Q.Rev.Biophys. 20 1.
- Zuber and Unwin (2013) Proc.Natl.Acad.Sci.U.S.A. 110 10622.
- Raftery et al. (1980) Science 208 1454.
- Karlin (2002) Nat.Rev.Neurosci. 3 102.
- Sine and Engel (2006) Nature 440 448.
- Bouzat (2012) J.Physiol.Paris 106 23.
- Grutter et al. (2004) Curr.Top.Med.Chem. 4 645.
- Smit et al. (2003) Ann.N.Y.Acad.Sci. 998 81.
- Hansen et al. (2005) EMBO J. 24 3635.
- Celie et al. (2005) J.Biol.Chem. 280 26457.
- Celie et al. (2004) Neuron 41 907.
- Rucktooa et al. (2009) Biochem.Pharmacol. 78 777.
- Li et al. (2011) Nat.Neurosci. 14 1253.
- Nayack and Auerbach (2013) Proc.Natl.Acad.Sci.U.S.A. 110 13654.
- Taly et al. (2009) Nat.Rev.Drug Discov. 8 733.
- Changeux et al. (2005) Science 308 1424.
- Taly et al. (2011) J.Mol.Graph.Model. 30 100.
- Lape et al. (2008) Nature 454 722.
- Mantione et al. (2012) Future Med.Chem. 4 2217.
- Ogden and Colquhoun (1985) Proc.R.Soc.Lond.B.Biol.Sci. 225 329.
- Bertrand and Gopalakrishnan (2007) Biochem.Pharmacol. 74 1155.
- Seo et al. (2009) J.Neurosci. 29 8734.
- Young et al. (2008) Proc.Natl.Acad.Sci.U.S.A. 105 14686.
- Lynagh and Lynch (2012) Trends Pharmacol.Sci. 33 432.
- Paton and Zaimis (1997) Br.J.Pharmacol. 120 60.
- Romano and Goldstein (1980) Science 210 647.
- Clarke et al. (1992) Trends Pharmacol.Sci. 13 407.
- Boulter et al. (1986) Nature 319 368.
- Harpsøe et al. (2013) J.Biol.Chem. 288 2559.
- Wang et al. (1996) J.Biol.Chem. 271 17656.
- Corringer et al. (2000) Annu.Rev.Pharmacol.Toxicol. 40 431.
- Kuryatov et al. (2008) Mol.Pharmacol. 74 132.
- Nelson et al. (2003) Mol.Pharmacol. 63 332.
- Srinivasan et al. (2011) J.Gen.Physiol. 137 59.
- Grishin et al. (2010) J.Biol.Chem. 285 22254.
- Krashia et al. (2010) PLoS One 5 e13611.
- Mazzo et al. (2013) J.Neurosci. 33 12316.
- Grupe et al. (2013) Br.J.Pharmacol. 168 2000.
- Millar (2008) Br.J.Pharmacol. 153 S177.
- Vallés et al. (2012) Biochim.Biophys.Acta. 1818 718.
- Rayes et al. (2009) J.Neurosci. 29 6022.
- Vetter et al. (2007) Proc.Natl.Acad.Sci.U.S.A. 104 20594.
- Elgoyhen et al. (2012) J.Physiol.Paris 106 47.
- Elgoyhen et al. (2001) Proc.Natl.Acad.Sci.U.S.A. 98 3501.
- Khiroug et al. (2002) J.Physiol. 540 425.
- Liu et al. (2009) J.Neurosci. 29 918.
- Murray et al. (2012) Mol.Pharmacol. 81 175.
- Holden-Dye et al. (2013) Parasitol.Int. 62 606.
- Millar and Denholm (2007) Invert.Neurosci. 7 53.
- Jones and Sattelle (2010) Adv.Exp.Med.Biol. 683 25.
- Matsuda et al. (2009) Mol.Pharmacol. 76 1.
- Blacquière et al. (2012) Ecotoxicology 21 973.
- Mao et al. (2006) Mol.Pharmacol. 70 1693. References
- David et al. (2010) Eur.J.Neurosci. 31 978.
- Genzen et al. (2001) J.Neurophysiol. 86 1773.
- Lips et al. (2002) Neuroscience 115 1.
- Hone et al. (2012) FASEB J. 26 917.
- Gotti et al. (2006) Trends Pharmacol.Sci. 27 482.
- Lukas et al. (1999) Pharmacol.Rev. 51 397.
- Wonnacott (1997) Trends Neurosci. 20 92.
- Mameli-Engvall et al. (2006) Neuron 50 911.
- Bürli et al. (2010) PLoS One 5 e11507.
- Dajas-Bailador and Wonnacott (2004) Trends Pharmacol.Sci. 25 317.
- Gu (2012) J.Neurosci. 32 12337.
- Jones et al. (1999) Trends Neurosci. 22 555.
- Klein and Yakel (2005) J.Physiol. 568 881.
- Wallace and Bertrand (2013) Biochem.Pharmacol. 85 1713.
- Broide and Leslie (1999) Mol.Neurobiol. 20 1.
- Poorhuis et al. (2013) J.Neurosci. 43 4843.
- Griguoli et al. (2013) J.Neurosci. 33 1044.
- Olincy and Freedman (2012) Handb.Exp.Pharmacol. 213 211.
- Mao et al. (2011) J.Neurosci. 31 6710.
- Zoli (1998) J.Neurosci. 18 4461.
- Stoker and Markou (2013) Curr.Opin.Neurobiol. 23 493.
- Orejarena et al. (2012) Neuropharmacology 63 235.
- Guillem et al. (2011) Science 333 888.
- Picard et al. (2013) Neuroimage 79 42.
- Hoda et al. (2008) Mol.Pharmacol. 74 379.
- Wada et al. (1990) Brain Res. 526 45.
- Flora et al. (2000) J.Neurochem. 75 18.
- Mao et al. (2008) J.Neurochem. 104 446.
- Berrettini and Doyle (2012) Mol.Psychiatry 17 856.
- Fowler et al. (2011) Nature 471 597.
- Frahm et al. (2011) Neuron 70 522.
- Livingstone and Wonnacott (2009) Biochem.Pharmacol. 78 744.
- Heath et al. (2010) Neuropsychopharmacology 35 2324.
- Exley et al. (2012) J.Neurosci. 32 2352.
- Gerzanich et al. (1998) J.Pharmacol.Exp.Ther. 286 311.
- Tapia et al. (2007) Mol.Pharmacol. 71 769.
- Chatterjee et al. (2013) PLoS One 8 e68300.
- Le Novère et al. (1996) Eur.J.Neurosci. 8 2428.
- Yang et al. (2009) Acta Pharmacol.Sin. 30 740.
- Salminen et al. (2007) Mol.Pharmacol. 71 1563.
- Kuryatov and Lindstrom (2011) Mol.Pharmacol. 79 126.
- Gotti et al. (2005) Mol.Pharmacol. 67 2007.
- Drenan et al. (2008) Mol.Pharmacol. 73 27.
- Dash and Lukas (2012) J.Biol.Chem. 287 14259.
- Broadbent et al. (2006) Mol.Pharmacol. 70 1350.
- Dash et al. (2011) J.Biol.Chem. 286 37976.
- Wada et al. (1989) J.Comp.Neurol. 284 314.
- Han et al. (2000) Eur.J.Neurosci. 12 3664.
- Lotfipour et al. (2013) J.Neurosci. 33 7728.
- Lamotte d’Incamps and Ascher (2013) Biochem.Pharmacol. 86 1114.
- Lipovsek et al. (2012) Proc.Natl.Acad.Sci.U.S.A. 109 4308.
- McIntosh et al. (2009) Biochem.Pharmacol. 78 693.
- Wessler and Kirkpatrick (2008) Br.J.Pharmacol. 154 1558.
- Schuller (2009) Nat.Rev.Cancer 9 195.
- Filippini et al. (2012) Curr.Drug Targets 13 644.
- Jensen et al. (2005) J.Med.Chem. 48 4705.
- Baker et al. (2004) Mol.Pharmacol. 65 453.
- Dunlop et al. (2007) Biochem.Pharmacol. 74 1172.
- Sidach et al. (2009) J.Neurosci.Methods 182 17.
- Malysz et al. (2009) Assay Drug Dev.Technol. 7 374.
- Papke et al. (2002) Br.J.Pharmacol. 137 49. 26 | tocris.com Tocris Scientific Review Series
- Jonsson et al. (2006) Anesthesiology 105 521.
- Verbitsky et al. (2000) Neuropharmacology 39 2515.
- Papke et al. (1996) Neurosci.Lett. 213 201.
- Alkondon et al. (1997) Eur.J.Neurosci. 9 2734.
- Alkondon and Albuquerque (2006) J.Pharmacol.Exp.Ther. 318 268.
- Wonnacott and Gallagher (2006) Marine Drugs 4 228.
- Swanson et al. (1986) Mol.Pharmacol. 29 250.
- Thomas et al. (1993) J.Neurochem. 60 2308.
- Feuerbach et al. (2005) Neuropharmacology 48 215.
- Stolerman et al. (1992) Neuropharmacology 31 311.
- MacPhail et al. (2007) Toxicology 234 83.
- Jarema et al. (2008) Neurotoxicol.Teratol. 30 220.
- Varanda et al. (1985) Mol.Pharmacol. 28 128.
- Jensen et al. (2003) Mol.Pharmacol. 64 865.
- Marks (2007) Neuropharmacology 53 390.
- Moroni et al. (2006) Mol.Pharmacol. 70 755.
- Tavares Xda (2012) J.Am.Chem.Soc. 134 11474.
- Pérez et al. (2012) Nat.Prod.Rep. 29 555.
- Coe et al. (2005) Bioorg.Med.Chem.Lett. 15 2974.
- Etter et al. (2008) Drug Alcohol Depend. 92 3.
- Mineur et al. (2007) Neuropharmacology 52 1256.
- Reavill et al. (1990) Neuropharmacology 29 619.
- Abin-Carriquiry et al. (2008) Eur.J.Pharmacol. 589 80.
- Ferger et al. (1998) Eur.J.Pharmacol. 360 155.
- Cunningham and McMahon (2011) Eur.J.Pharmacol. 654 47.
- Smith et al. (2007) Psychopharmacology (Berl). 190 157.
- Jutkiewicz et al. (2011) J.Pharmacol.Exp.Ther. 339 194.
- Badio and Daly (1994) Mol.Pharmacol. 45 563.
- Xiao and Kellar (2004) J.Pharmacol.Exp.Ther. 310 98.
- Gerzanich et al. (1995) Mol.Pharmacol. 48 774.
- Alkondon and Albuquerque (1995) J.Pharmacol.Exp.Ther. 274 771.
- Damaj et al. (1998) J.Pharmacol.Exp.Ther. 284 1058.
- Khan et al. (1998) Neuropharmacology 37 1515.
- Cucchiaro et al. (2006) Neuropharmacology 50 769.
- Ganesh et al. (2008) Eur.J.Pharmacol. 584 93.
- Marubio et al. (1999) Nature 398 805.
- Benowitz et al. (2009) Handb.Exp.Pharmacol. 192 29.
- Matta et al. (2007) Psychopharmacology (Berl). 190 269.
- Buccafusco and Terry Jr (2003) Life Sci. 72 2931.
- O’Leary et al. (2008) J.Pharmacol.Exp.Ther. 325 646.
- Oliver et al. (2007) Vaccine 25 7354.
- Bitner et al. (2007) J.Neurosci. 27 10578.
- Thomsen et al. (2008) Neuroscience 154 741.
- Thomsen et al. (2011) PLoS One 6 e27014.
- Kohlhaas et al. (2012) Psychopharmacology (Berl). 220 823.
- Briggs et al. (2008) Br.J.Pharmacol. 153 1054.
- Schrimpf et al. (2012) Bioorg.Med.Chem.Lett. 22 1633.
- Toyohara et al. (2010) PLoS One 5 e8961.
- Garcha et al. (1993) Psychopharmacology (Berl). 110 347.
- Binns and Salt (2000) Vis.Neurosci. 17 283.
- Spivak et al. (1986) Eur.J.Pharmacol. 120 127.
- Manetti et al. (1999) Bioorg.Med.Chem.Lett. 7 457.
- Dewey and Xie (2013) Phytochemistry 94 10.
- Marks et al. (1986) Mol.Pharmacol. 30 427.
- Stolerman et al. (1995) Psychopharmacology (Berl). 117 430.
- Mastropaolo et al. (2004) Behav.Brain Res. 153 419.
- Welch et al. (2013) Toxicol.Appl.Pharmacol. 266 366.
- Mullen et al. (2000) J.Med.Chem. 43 4045.
- Papke et al. (2004) Bioorg.Med.Chem.Lett. 14 1849.
- Levin et al. (1999) Behav.Pharmacol. 10 675.
- Van Kampen et al. (2004) Psychopharmacology (Berl). 172 375.
- Grottick et al. (2003) Neuropsychopharmacology 28 880.
- Imming (2001) Eur.J.Med.Chem. 36 375.
- Abin-Carriquiry et al. (2006) Eur.J.Pharmacol. 536 1.
- Houlihan et al. (2001) J.Neurochem. 78 1029.
- Abin-Carriquiry et al. (2010) Eur.J.Pharmacol. 634 89.
- Kem et al. (1997) J.Pharmacol.Exp.Ther. 283 979.
- Arendash et al. (1995) Brain Res. 674 252.
- Stevens et al. (1998) Psychopharmacology (Berl). 136 320.
- Mukhin et al. (2000) Mol.Pharmacol. 57 642.
- Mogg et al. (2004) Neuropharmacology 47 848.
- Kennett et al. (2012) Nicotine Tob.Res. 14 1339.
- Wildeboer and Stevens (2008) Brain Res. 1224 29.
- Huang et al. (2011) Neuropharmacology 60 861.
- Cavanagh et al. (2010) Synapse 64 111.
- Miller et al. (2000) Neuropharmacology 39 2654.
- Dwoskin and Crooks (2001) J.Pharmacol.Exp.Ther. 298 395.
- Kaniaková et al. (2011) Eur.J.Pharmacol. 658 108.
- Farook et al. (2009) Physiol.Behav. 97 503.
- Stead and Hughes (2012) Cochrane Database Syst.Rev. 2 CD000124.
- Crooks et al. (2011) Curr.Top.Med.Chem. 11 1103.
- Miller et al. (2007) Dug Alcohol Depend. 89 282.
- Jeong et al. (2010) Pflugers Arch. 460 851.
- Levin and Christopher (2003) Pharmacol.Biochem.Behav. 76 133.
- Roni and Rahman (2013) Prog.Neuropsychopharmacol.Biol.Psychiatry 41 44.
- Wishka et al. (2006) J.Med.Chem. 49 4425.
- Kita et al. (2013) Psychopharmacology (Berl). 225 543.
- Shilliday et al. (2010) Drug Metab.Lett. 4 162.
- Bodnar et al. (2005) J.Med.Chem. 48 905.
- Hajós et al. (2005) J.Pharmacol.Exp.Ther. 312 1213.
- Pandya and Yakel (2013) Neuropharmacology 70 35.
- Young et al. (2013) Behav.Brain Res. 240 119.
- Brunzell and McIntosh (2012) Neuropsychopharmacology 37 1134.
- Igari et al. (2014) Neuropsychopharmacology 39 445.
- Turcanu et al. (2012) Neurosci.Lett. 516 212.
- Bencherif et al. (1996) J.Pharmacol.Exp.Ther. 279 1413.
- Papke (2000) J.Neurochem. 75 204.
- Garduño et al. (2012) J.Neurosci. 32 15148.
- Lippiello et al. (1996) J.Pharmacol.Exp.Ther. 279 1422.
- Damaj et al. (1999) J.Pharmacol.Exp.Ther. 291 390.
- Rowley et al. (2008) Anesth.Analg. 107 1052.
- Bencherif et al. (1998) J.Pharmacol.Exp.Ther. 284 886.
- Lopez-Hernandez et al. (2007) Neuropharmacology 53 134.
- Beracochea et al. (2008) Psychopharmacology (Berl). 196 555.
- Wang et al. (2009) J.Neurosci. 29 10961.
- Wang et al. (2010) Biol.Psychiatry 67 522.
- Xiao et al. (2006) Mol.Pharmacol. 70 1454.
- Zwart et al. (2008) Mol.Pharmacol. 73 1838.
- Liu et al. (2013) J.Med.Chem. 56 3000.
- Rezvani et al. (2010) Psychopharmacology (Berl). 211 161.
- Turner et al. (2013) Neuropsychopharmacology 38 2035.
- Roncarati et al. (2009) J.Pharmacol.Exp.Ther. 329 459.
- Haydar et al. (2009) Bioorg.Med.Chem. 17 5247.
- Marrero et al. (2004) J.Pharmacol.Exp.Ther. 309 16.
- Bencherif et al. (2000) Eur.J.Pharmacol. 409 45.
- Chen et al. (2003) Neuropharmacology 45 334.
- Richardson et al. (1985) Nature 316 126.
- Rothlin (2003) Mol.Pharmacol. 63 1067.
- Cui et al. (2009) Eur.J.Pharmacol. 609 74.
- Kohnomi et al. (2010) Brain Res. 1353 152.
- Sharples et al. (2000) J.Neurosci. 20 2783.
- Cao et al. (2005) Neuropharmacology 48 72.
- Mihalak et al. (2006) Mol.Pharmacol. 70 801.
- Papke et al. (2010) J.Pharmacol.Exp.Ther. 333 501.
- Bordia et al. (2012) J.Pharmacol.Exp.Ther. 342 327.
- Hendrickson et al. (2010) J.Neurosci. 30 10169.
- Rollema et al. (2007) Neuropharmacology 52 985.
- Wildeboer-Andrud and Stevens (2011) Pharmacol.Biochem.Behav. 100 17. tocris.com | 27 Nicotinic ACh Receptors
- Obach et al. (2006) Drug Metab.Dispos. 34 121.
- Roegge et al. (2006) Nicotinic receptor antagonists in rats. In: Levin and Buccafusco ed. Animal Models of Cognitive Impairment. CRC Press, Boca Raton, FL.
- Bertrand et al. (1990) Proc.Natl.Acad.Sci.U.S.A. 87 1993.
- Bowman (1958) Br.J.Pharmacol.Chemother. 13 521.
- Christ and Blaber (1968) J.Pharmacol.Exp.Ther. 160 159.
- Lukas (1989) J.Pharmacol.Exp.Ther. 251 175.
- Walker et al. (1993) Comp.Biochem.Physiol.C. 106 49.
- Bini et al. (2006) J.Exp.Zool.A.Comp.Exp.Biol. 305 472.
- Pereira et al. (1993) J.Pharmacol.Exp.Ther. 265 1474.
- Dwoskin et al. (2004) Bioorg.Med.Chem.Lett. 14 1863.
- Dwoskin et al. (2008) J.Pharmacol.Exp.Ther. 326 563.
- Rahman et al. (2008) Br.J.Pharmacol. 153 792.
- Papke et al. (2008) Neuropharmacology 54 1189.
- Wooters et al. (2011) Br.J.Pharmacol. 163 346.
- Fuchs et al. (2003) Ann.N.Y.Acad.Sci. 998 93.
- Dellisanti et al. (2007) Nat.Neurosci. 10 1222.
- Huang et al. (2013) Biochem.J. 454 303.
- Wu et al. (2004) J.Pharmacol.Exp.Ther. 311 80.
- Ng et al. (2007) Proc.Natl.Acad.Sci.U.S.A. 104 8059.
- Shahsavar et al. (2012) PLoS One 7 e40757.
- Harvey et al. (1996) J.Neurochem. 67 1953.
- Capelli et al. (2011) Br.J.Pharmacol. 163 313.
- Soll et al. (2013) Neuropharmacology 73 19.
- Jutkiewicz et al. (2011) J.Pharmacol.Exp.Ther. 339 194.
- Guy and Fletcher (2013) Psychopharmacology (Berl). 225 429.
- André et al. (2011) Neuropharmacology 60 617.
- Jackson et al. (2013) Neuropharmacology 70 228.
- Struthers et al. (2009) Pharmacol.Biochem.Behav. 94 319.
- Ward et al. (1990) FEBS Lett. 270 45.
- Dupuis et al. (2011) J.Neurophysiol. 106 1604.
- Mogg et al. (2002) J.Pharmacol.Exp.Ther. 302 197.
- Feng et al. (2011) Behav.Brain Res. 220 100.
- Lockman et al. (2005) J.Neurochem. 94 37.
- van Kampen et al. (2004) Psychopharmacology (Berl). 172 375.
- Gotti et al. (1998) Br.J.Pharmacol. 124 1197.
- Maggi et al. (1999) Br.J.Pharmacol. 126 285.
- Xu et al. (2010) Respir.Res. 11 13.
- Brown et al. (2012) Angiogenesis 15 99.
- Cao et al. (2005) J.Pharmacol.Exp.Ther. 312 1298.
- Tanibuchi (2010) Brain Res. 1348 200.
- Buckett (1968) Ir.J.Med.Sci. 7 565.
- Wenningmann and Dilger (2001) Mol.Pharmacol. 60 790.
- Paul et al. (2002) Anesth.Analg. 94 597.
- Jonsson Fagerlund et al. (2009) Anesthesiology 110 1244.
- Yost and Winegar (1997) Cell Mol.Neurobiol. 17 35.
- Liu and Dilger (2009) Mol.Pharmacol. 75 166.
- Zaveri et al. (2010) J.Med.Chem. 53 8187.
- Lee (2005) J.R.Coll.Physicians Edinb. 35 83.
- Wang et al. (2003) J.Biol.Chem. 278 32284.
- Bertrand et al. (1992) Neurosci.Lett. 146 87.
- Brams et al. (2011) PLoS Biol. 9 e1001034.
- McMahon and Kauer (1997) J.Neurophysiol. 78 2493.
- Brown et al. (1998) J.Physiol. 507 653.
- Yan et al. (2006) Mol.Pharmacol. 70 571.
- Schwartz and Mindlin (1988) J.Pharmacol.Exp.Ther. 244 963.
- Wotring and Yoon (1995) Neuroscience 67 293.
- Azam and McIntosh (2009) Acta Pharmacol.Sin. 30 771.
- Armishaw (2010) Toxins (Basel) 2 1471.
- Vincler et al. (2006) Proc.Natl.Acad.Sci.U.S.A. 103 17880.
- Sandall et al. (2003) Biochemistry 42 6904.
- Clark et al. (2005) Proc.Natl.Acad.Sci.U.S.A. 102 13767.
- Satkunanathan et al. (2005) Brain Res. 1059 149.
- van Lierop et al. (2013) ACS Chem.Biol. 8 1815.
- Nevin et al. (2007) Mol.Pharmacol. 72 1406.
- Callaghan et al. (2008) J.Neurosci. 28 10943.
- Adams et al. (2012) Br.J.Pharmacol. 166 486.
- Napier et al. (2012) Neuropharmacology 62 2202.
- Klimis et al. (2011) Pain 152 259.
- Martinez et al. (1995) Biochemistry 34 14519.
- Park et al. (2001) J.Biol.Chem. 276 49028.
- López-Vera et al. (2007) FEBS J. 274 3972.
- McIntosh et al. (1994) J.Biol.Chem. 269 16733.
- Johnson et al. (1995) Mol.Pharmacol. 48 194.
- Broxton et al. (1999) J.Neurochem. 72 1656.
- Ellison et al. (2004) Biochemistry 43 16019.
- Yu et al. (2011) PLoS Comput.Biol. 7 e1002011.
- Pereira et al. (1996) J.Pharmacol.Exp.Ther. 278 1472.
- Barik and Wonnacott (2006) Mol.Pharmacol. 69 618.
- Cartier et al. (1996) J.Biol.Chem. 271 7522.
- Champtiaux et al. (2002) J.Neurosci. 15 1208.
- McIntosh et al. (2004) Mol.Pharmacol. 65 944.
- Kulak et al. (2002) Mol.Pharmacol. 61 230.
- Exley et al. (2013) Eur.J.Neurosci. [Epub ahead of print].
- McCallum et al. (2005) Mol.Pharmacol. 68 737.
- Löf et al. (2007) Psychopharmacology (Berl). 195 333.
- Brunzell et al. (2010) Neuropsychopharmacology 35 665.
- Gotti et al. (2010) J.Neurosci. 30 5311.
- Okada et al. (2008) Eur.J.Pharmacol. 587 322.
- Dowell et al. (2003) J.Neurosci. 23 8445.
- Azam and McIntosh (2005) J.Pharmacol.Exp.Ther. 312 231.
- Yang et al. (2011) J.Neurosci. 31 2537.
- Fainzilber et al. (1994) Biochemistry 33 9523.
- Luo et al. (1999) Biochemistry 38 14542.
- Everhart et al. (2003) J.Pharmacol.Exp.Ther. 306 664.
- Hogg et al. (2003) J.Biol.Chem. 278 26908.
- Luo et al. (1998) J.Neurosci. 18 8571. 347. Kulak et al. (2001) J.Neurochem. 77 1581.
- Grady et al. (2001) J.Neurochem. 76 258.
- Park et al. (2006) Pflugers Arch. 452 775.
- Glick et al. (2011) Eur.J.Pharmacol. 669 71.
- McCallum et al. (2012) Neuropharmacology 63 434.
- Herrero et al. (2002) J.Neurosci. 22 377.
- Peng et al. (2013) Mol.Pharmacol. 84 459.
- Mahata et al. (2010) Regul.Pept. 162 33.
- Yang et al. (2013) Proc.Natl.Acad.Sci.U.S.A. 110 12078.
- Sahu et al. (2012) J.Cell.Sci. 125 2323.
- Plummer et al. (1955) J.Pharmacol.Exp.Ther. 115 172.
- Reuben et al. (1998) Br.J.Pharmacol. 125 1218.
- Costa et al. (2001) Brain Res. 888 336.
- Hunter (1950) Lancet 1 251.
- Ascher et al. (1979) J.Physiol. 295 139.
- DeNoble and Mele (2006) Psychopharmacology (Berl). 184 266.
- Ortiz et al. (2012) Nicotine Tob.Res. 14 711.
- Benwell et al. (1995) Br.J.Pharmacol. 114 454.
- Malin et al. (1997) Pharmacol.Biochem.Behav. 58 695.
- Marks et al. (1999) J.Pharmacol.Exp.Ther. 289 1090.
- Eaton et al. (2003) Mol.Pharmacol. 64 1283.
- Ford et al. (1956) Am.J.Med.Sci. 232 129.
- Bacher et al. (2009) Expert Opin.Pharmacother. 10 2709.
- Arias et al. (2010) Int.J.Biochem.Cell Biol. 42 1007.
- Papke et al. (2001) J.Pharmacol.Exp.Ther. 297 646.
- Yalcin et al. (2009) J.Physiol.Pharmacol. 60 133.
- Francis et al. (1998) Biophys.J. 74 2306.
- Papke et al. (2005) Mol.Pharmacol. 67 1977.
- Damaj et al. (2005) Eur.J.Pharmacol. 521 43. 28 | tocris.com Tocris Scientific Review Series
- Rehni and Singh (2010) Naunyn Schmiedebergs Arch.Pharmacol. 382 279.
- Fryer and Lukas (1999) J.Pharmacol.Exp.Ther. 288 88.
- Slemmer et al. (2000) J.Pharmacol.Exp.Ther. 295 321.
- Sidhpura (2007) Eur.J.Pharmacol. 567 102.
- Arias (2009) Int.J.Biochem.Cell Biol. 41 2098.
- Damaj et al. (2004) Mol.Pharmacol. 66 675.
- Pandhare et al. (2012) Biochemistry 51 2425.
- García-Colunga et al. (1997) Proc.Natl.Acad.Sci.U.S.A. 94 2041.
- Feuerbach et al. (2005) Neuropharmacology 48 215.
- Weber et al. (2013) Neuropharmacology 72 88.
- Grinevich et al. (2009) Neuropharmacology 57 183.
- Zheng et al. (2010) J.Pharmacol.Exp.Ther. 335 401.
- Villarroya et al. (1997) Eur.J.Pharmacol. 320 249.
- Herrero et al. (1999) Br.J.Pharmacol. 127 1375.
- Ween et al. (2010) Neurochem.Int. 57 269.
- Huettner et al. (1988) Proc.Natl.Acad.Sci.U.S.A. 85 1307.
- Briggs and McKenna (1996) Neuropharmacology 35 407.
- Buisson and Bertrand (1998) Mol.Pharmacol. 53 555.
- Amador and Dani (1991) Synapse 7 207.
- Ho and Flood (2004) Anesthesiology 100 657.
- Aracava et al. (2005) J.Pharmacol.Exp.Ther. 312 1195.
- Lee et al. (2012) PLoS One 7 e40326.
- Hilmas et al. (2001) J.Neurosci. 21 7463.
- Albuquerque and Schwarcz (2013) Biochem.Pharmacol. 85 1027.
- Stone and Darlington (2013) Br.J.Pharmacol. 169 1211.
- Valera et al. (1992) Proc.Natl.Acad.Sci.U.S.A. 89 9949.
- Ke and Lukas (1996) J.Neurochem. 67 1100.
- Garbus et al. (2001) Neuroreport 12 227.
- Fernandez-Ballester et al. (1994) Biochem.Soc.Trans. 22 776.
- Gangitano et al. (2009) Genes Brain Behav. 8 398.
- García-Colunga and Miledi (1999) Proc.Natl.Acad.Sci.U.S.A. 96 4113.
- Matsubayashi et al. (1998) J.Pharmacol.Exp.Ther. 284 904.
- Parri et al. (2011) Biochem.Pharmacol. 82 931.
- Liu et al. (2001) Proc.Natl.Acad.Sci.U.S.A. 98 4734.
- Grassi et al. (2003) J.Physiol. 547 147.
- Mehta et al. (2009) J.Neurochem. 109 1452.
- Snyder et al. (2005) Nat.Neurosci. 8 1051.
- Abbott et al. (2008) Neurobiol.Aging 29 992.
- Puzzo and Arancio (2013) J.Alzheimers Dis. 33 S111.
- Talantova et al. (2013) Proc.Natl.Acad.Sci.U.S.A. 110 E2518.
- Grönlien et al. (2007) Mol.Pharmacol. 72 715.
- Schrattenholz et al. (1996) Mol.Pharmacol. 49 1.
- Chimienti et al. (2003) Hum.Mol.Genet. 12 3017.
- Kawashima et al. (2012) Ann.N.Y.Acad.Sci. 1261 7.
- Faghih et al. (2007) Recent Pat.CNS Drug Discov. 2 99.
- Sattelle et al. (2009) Biochem.Pharmacol. 78 836.
- Dey and Chen (2011) J.Biomol.Struct.Dyn. 28 695.
- Collins et al. (2011) Neuropharmacology 61 1306.
- Freitas et al. (2013) J.Pharmacol.Exp.Ther. 344 264.
- Dickinson et al. (2007) J.Neurochem. 100 1089.
- Hu et al. (2009) Br.J.Pharmacol. 158 1857.
- daCosta et al. (2011) J.Neurosci. 31 13870.
- Gill et al. (2011) Proc.Natl.Acad.Sci.U.S.A. 108 5867.
- Bertrand et al. (2008) Mol.Pharmacol. 74 1407.
- Luttmann et al. (2009) ChemMedChem 4 1874.
- Hamouda et al. (2013) J.Neurosci. 33 485.
- Moroni et al. (2008) J.Neurosci. 28 6884.
- Kuryatov et al. (2000) Neuropharmacology 39 2570.
- Faghih et al. (2009) J.Med.Chem. 52 3377.
- Malysz et al. (2009) J.Pharmacol.Exp.Ther. 330 257.
- Anderson et al. (2008) J.Pharmacol.Exp.Ther. 324 179.
- Peters et al. (2004) Planta Med. 70 883.
- Sala et al. (2005) Neurosci.Lett. 373 144.
- Kim et al. (2007) Bioorg.Med.Chem.Lett. 17 4855.
- Weltzin and Schulte (2010) J.Pharmacol.Exp.Ther. 334 917.
- German et al. (2011) J.Med.Chem. 54 7259.
- Liu (2013) Psychopharmacology (Berl). 230 203.
- Samochocki et al. (2003) J.Pharmacol.Exp.Ther. 305 1024.
- Dajas-Bailador et al. (2003) Mol.Pharmacol. 64 1217.
- Akk and Steinbach (2005) J.Neurosci. 25 1992.
- Smith et al. (2009) Biochem.Pharmacol. 78 889.
- Storch et al. (1995) Eur.J.Pharmacol. 290 207.
- Militante et al. (2008) Mol.Pharmacol. 74 764.
- Ludwig et al. (2010) J.Recept.Signal Transduct.Res. 30 469.
- Krause et al. (1998) Mol.Pharmacol. 53 283.
- Collins and Millar (2010) Mol.Pharmacol. 78 198.
- Larocque and Hoffman (2012) Clin.Toxicol.(Phila). 50 231
- Levandoski et al. (2003) Eur.J.Pharmacol. 471 9.
- Broad et al. (2006) J.Pharmacol.Exp.Ther. 318 1108.
- Timmermann et al. (2007) J.Pharmacol.Exp.Ther. 323 294.
- Hurst et al. (2005) J.Neurosci. 25 4396.
- Pandya and Yakel (2013) Biochem.Pharmacol. 86 1054.
- El Kouhen et al. (2009) Br.J.Pharmacol. 156 638.
- Grybko et al. (2011) Eur.J.Neurosci. 33 1786.
- Shen and Yakel (2012) J.Mol.Neurosci. 48 14.
- daCosta and Sine (2013) Proc.Natl.Acad.Sci.U.S.A. 110 6595.
- McLean et al. (2012) J.Psychopharmacol. 26 1265.
- Kalappa et al. (2013) Br.J.Pharmacol. 169 1862.
- Munro et al. (2012) Br.J.Pharmacol. 167 421.
- Livingstone et al. (2009) Eur.J.Neurosci. 29 539.
- Christensen et al. (2010) J.Neurochem. 114 1205.
- Gill et al. (2013) PLoS One 8 e55047.
- Le Novère et al. (2002) J.Neurobiol. 53 447.
- Filevich et al. (2010) J.Inorg.Biochem. 104 1248.
- Absalom et al. (2013) J.Biol.Chem. 288 26521.
- Srinivasan et al. (2012) Int.J.Mol.Sci. 13 10022.
- Tochitsky et al. (2012) Nature Chem. 10 105.
- Brust et al. (2012) Curr.Drug Targets 13 594.
- Wong et al. (2013) J.Nucl.Med. 54 1308.
- Moser et al. (2007) J.Neurochem. 102 479.
- Chang and Fischbach (2006) J.Neurosci. 26 11295.
- Shelukhina et al. (2009) J.Neurochem. 109 1087.
- Hone et al. (2013) J.Neurochem. 114 994.
- Gomez-Varela and Berg (2013) J.Neurosci. 33 17062.
- Sekine-Aizawa and Huganir (2004) Proc.Natl.Acad.Sci.U.S.A. 101 17114.
- Hannan et al. (2013) Methods Enzymol. 521 109.
- Justinova et al. (2013) Nature Neurosci. 16 1652.
- Caffery et al. (2009) Eur.J.Neurosci. 30 2064.
- Woodruff-Pak et al. (1994) Brain Res. 645 309.
- Kem (2000) Behav. Brain Res. 113 169.
- Hibbs et al. (2009) EMBO J. 28 3040.
- Stokes et al. (2004) Mol.Pharmacol. 66 14.
- Mahnir et al. (1998) Biopharm.Drug Dispos. 19 147.
- Callahan et al. (2014) Psychopharmacology (Berl). [Epub ahead of print].
- Kem et al. (2004) Mol.Pharmacol. 65 56.
About Tocris Bioscience
Tocris Bioscience is your trusted supplier of high-performance life science reagents, including receptor agonists & antagonists, enzyme inhibitors, ion channel modulators, fluorescent probes & dyes, and compound libraries. Our catalog consists of over 4,500 research tools, covering over 400 protein targets enabling you to investigate and modulate the activity of numerous signaling pathways and physiological processes.
We have been working with scientists for over 30 years to provide the life science community with research standards, as well as novel and innovative research tools. We understand the need for researchers to trust their research reagents, which is why we are committed to supplying our customers with the highest quality products available, so you can publish with confidence.
Tocris is part of the protein sciences division of Bio-Techne, which also includes the best in class brands R&D Systems, Novus Biologicals, ProteinSimple, and Advanced Cell Diagnostics. Bio-Techne has united these brands to provide researchers with a full portfolio of research reagents, assays, and protein platforms. For more information on Bio-Techne and its brands, please visit bio-techne.com.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices, and treatments.