After Alzheimer’s Disease, the second most prevalent neurodegenerative disease is Parkinson or Parkinson’s Disease (PD). The diagnosis of this disease is based on the occurrence of bradykinesia, unilateral or asymmetric resting tremor, and rigidity. These motor features are caused when dopaminergic neurons degenerate in the substantia nigra pars compacta (SNpc).
In addition, neurodegeneration occurs in non-dopaminergic pathways and leads a string of non-motor features, including autonomic dysfunction, sleep disorders, and cognitive impairment. The factors that lead to PD include multiple different mutations in genes encoding proteins such as LRRK2, α-synuclein, PINK1, and Parkin, with mutations in glucocerebrosidase (GBA) present the highest risk for the development of PD.
Environmental and Genetic Factors
There has been more and more evidence that genetics has a principal role to play in the etiology of PD. A number of individual gene mutations are related to autosomal dominant or recessive PD. Together, these mutations account for 10%–15% of PD cases. The most common genetic cause of PD are LRRK2 mutations, which are observed in 2%–3% of familial cases and 0.5%–1.0% of all cases identified in the UK.
The most prevalent cause of early-onset (within the age of 30 years) PD are Parkin mutations. Several association loci—such as GBA and tau, as well as genes in lysosomal, mitochondrial and inflammatory pathways—have been identified by genome-wide association studies; for instance, mutations in Parkin and PINK1 genes lead to mitochondrial dysfunction, a significant feature of PD pathogenesis.
In terms of numbers, GBA mutations constitute the most significant risk factor for PD. It has been predicted that in the United Kingdom, GBA mutations are found in 7%–10% of PD patients. On the other hand, although PD is not caused by any environmental factor, it has been observed that pesticide exposure, rural living, and some toxins increase PD risk by two- to three-fold, while coffee intake and cigarette smoking reduce the risk.
Disruption of Neuronal Pathways
Degeneration of the dopaminergic systems alone are not responsible for all PD symptoms; noradrenaline, serotonin, GABA (not shown), and acetylcholine (not shown) pathways are also acutely affected (Figure 1). It has been found that Lewy bodies occur early in the lower brain stem and olfactory bulb without any loss in neuronal cells. As the disease turns symptomatic, evidence of dopaminergic cell loss in the SNpc and Lewy-body deposition emerge. Other brain stem nuclei—for instance, substantia innominata and locus coeruleus— are also affected by the degenerative process. Noticeable non-dopaminergic features can be observed in highly advanced cases of PD due to neuron loss in the brainstem, subcortex, and cortex, as well as in peripheral autonomic sites.
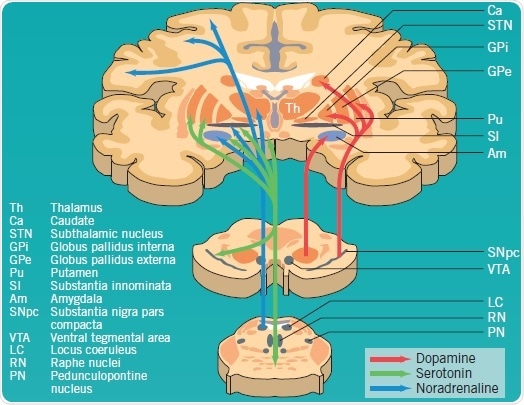
Figure 1: Dopaminergic, serotonergic and noradrenergic pathways affected by PD pathology. Image credit: Tocris Bioscience
In PD pathogenesis, the complex direct and indirect pathways of the basal ganglia are disrupted (Figure 2). The SNpc’s dopaminergic neurons project toward GABA neurons in the striatum, and are inhibitory (GABA-enk) or excitatory (GABA-SP). GABA-SP projections of inhibitory synapses to the GPi are involved in the direct pathway. The SNpr is a functional component of the GPi. In the indirect pathway, GABA-enk inhibitory projections are observed toward the GPe and there is onward inhibitory input onto the STN glutamatergic (Glu) neurons. The STN provides excitatory input into the GPi, but presumably also into the SNpc.
In PD, apart from the loss of dopaminergic neurons in the SNpc, dopamine levels in the striatum are observed to decline, which results in reduced activity of GABA-SP and increased activity of GABA-enk. This consequently enhances the activation of the STN’s glutamatergic excitatory output and, hence, that of the GPi with the ensuing inhibition of the thalamus and its cortical projections.
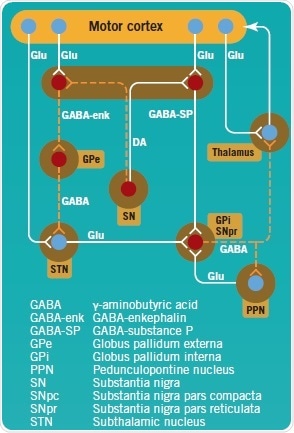
Figure 2: Direct and indirect pathways of the basal ganglia, projecting to and controling the motor cortex. Image credit: Tocris Bioscience
Current and Emerging Treatments for PD
The motor features of PD are caused by the loss of dopaminergic pathways, particularly the nigrostriatal pathway. Normal dopamine tone is disrupted and the function of basal ganglia is impaired due to the loss of dopamine neurons. Symptoms can be improved by reducing glutamatergic or cholinergic stimulation, or by increasing dopamine stimulation (Figure 3).
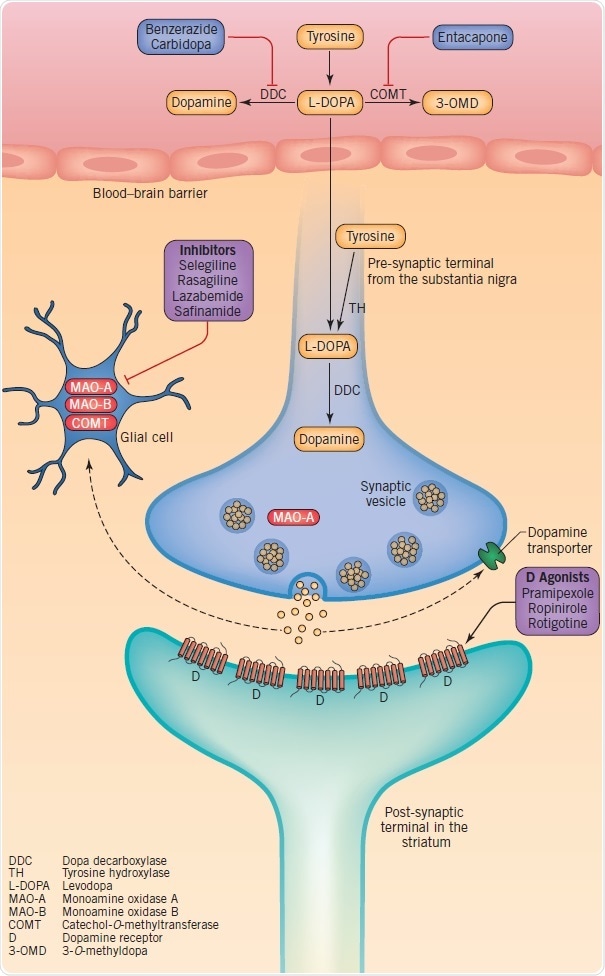
Figure 3: Therapeutic targets at dopaminergic synapses, including dopamine synthesis, metabolism, storage and synaptic transmission. Image credit: Tocris Bioscience
Dopamine synthesis and catabolism offer the basis for drug therapies targeted at the symptomatic treatment of motor symptoms. Dopamine is produced when tyrosine is converted into levodopa by tyrosine hydroxylase (TH) with the subsequent decarboxylation of levodopa through dopa decarboxylase (DDC). Glial monoamine oxidase (MAO)-A and MAO-B, and intraneuronal MAO-A are responsible for the metabolization of dopamine. Levodopa is used in dopamine-replacement therapy since dopamine does not cross the blood–brain barrier (BBB). As soon as levodopa crosses into the brain the terminals of the surviving nigrostriatal neurons, and possibly microglia and serotonergic neurons, convert it into dopamine.
Dopamine is stored in vesicles and discharged into the synaptic cleft as a reaction to physiological stimuli. The discharged dopamine gets binds to dopaminergic receptors and can subsequently be absorbed again into the pre-synaptic terminal by the dopamine transporter (DAT), or metabolized by catechol-O-methyltransferase (COMT) and MAO. Based on their specific profile, dopamine agonists tend to activate pre- and post-synaptic dopamine D1, D2, and D3 receptors. Upon oral administration, they get absorbed and cross the BBB. Since MAO-B inhibitors minimize the disintegration of dopamine, they increase its synaptic half-life and hence the amount re-absorbed into the pre-synaptic terminal. Although COMT inhibitors are orally active, they function in the intestines to minimize levodopa’s peripheral metabolism and improve its penetration into the central nervous system.
Despite providing the most symptomatic relief, levodopa causes long-term complications such as dyskinesias (involuntary movements), and over time the effects of a dose wears off. Based on their clinical profile, patients may be started on levodopa, a MAO-B inhibitor, or a dopamine agonist. Unavoidably, all PD patients will have to be given levodopa; moreover, this is now being used more and more together with a COMT inhibitor. Sadly, none of these therapies have been established to delay the advancement of the disease or the occurrence of non-motor, principally non-dopaminergic features.
Better insights into the pathogenesis and etiology of Parkinson’s disease have unraveled a number of key pathways that have turned out to be targets for prospective treatments. Although there are already therapeutic approaches for relieving the symptomatic stages of PD, new genetic understanding might enable the use of preventative, neuroprotective treatments for those at risk of developing PD, thereby slowing down the onset and advancement of the disease. Along with efforts of prevention and control of symptomatic PD, scientists have also turned toward the use of stem cells to replace the diseased neurons.
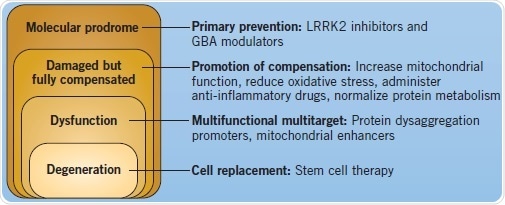
Figure 4: Disease course and different therapeutic interventions for PD. Image credit: Tocris Bioscience
Pathways for Potential Intervention in Aberrant α-Synuclein Metabolism
Lysosomal dysfunction is regarded as a key part of PD pathology, specifically because α-synuclein is mainly turned over by chaperone-mediated autophagy. In addition, neurodegeneration in PD has been associated with the development of toxic protein aggregates, for example, those that develop due to the conversion of α-helix protein structures into β-sheet configurations. Accumulation of α-synuclein oligomers occurs if there is a defect in this pathway, promoting the formation of aggregates. Further support for the significance of lysosomal dysfunction in PD comes from the involvement of LRRK2 in autophagy, and the association of GBA mutations.
The is great interest in α-synuclein toxic oligomers formation and their interneuronal propagation and aggregate formation. This feature of PD has been equated to prion disorders. As illustrated in Figure 5, a number of therapeutic approaches have been suggested to decrease the effects of aberrant α-synuclein metabolism in PD.
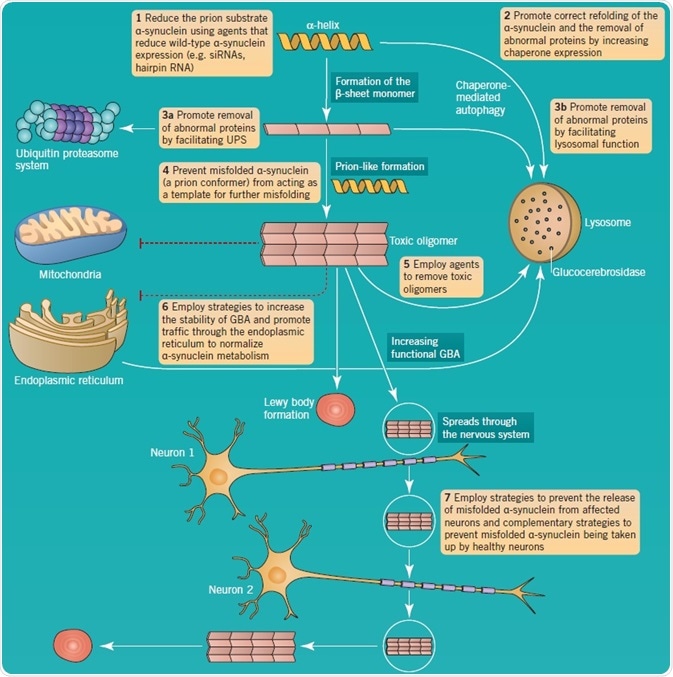
Figure 5: α-synuclein aggregation and pathology as a therapeutic target in PD. Image credit: Tocris Bioscience
References
- Schapira et al. (2014) The Lancet 384 545
- Olanow and Schapira (2013) Ann. Neurol. 74 337
About Tocris Bioscience
Tocris Bioscience is your trusted supplier of high-performance life science reagents, including receptor agonists & antagonists, enzyme inhibitors, ion channel modulators, fluorescent probes & dyes, and compound libraries. Our catalog consists of over 4,500 research tools, covering over 400 protein targets enabling you to investigate and modulate the activity of numerous signaling pathways and physiological processes.
We have been working with scientists for over 30 years to provide the life science community with research standards, as well as novel and innovative research tools. We understand the need for researchers to trust their research reagents, which is why we are committed to supplying our customers with the highest quality products available, so you can publish with confidence.
Tocris is part of the protein sciences division of Bio-Techne, which also includes the best in class brands R&D Systems, Novus Biologicals, ProteinSimple, and Advanced Cell Diagnostics. Bio-Techne has united these brands to provide researchers with a full portfolio of research reagents, assays, and protein platforms. For more information on Bio-Techne and its brands, please visit bio-techne.com.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices, and treatments.