Introduction
Understanding neurite dynamics is essential to study neuronal differentiation, embryonic development, neuronal injury and regeneration, and neuropathological disorders.
Neurite dynamic measurements are regularly applied as a screening assay in the development of neurotherapeutic drugs and neurotoxicity triggered by an environmental chemical or drug can be predicted from the changes in neurite length.
Concatenation of neurite length and branch point measurements taken at single points in time is the widely used approach for investigating neurite dynamics. In this method, neuronal cultures are grown and exposed to a treatment condition, and then the experiment is halted with fixation and immunostaining steps.
The next step is performing image acquisition and analysis. Although this approach plays a major role in neurobiology, it is laborious and provides only a single timepoint of data, which is inadequate to reveal the dynamic effects of the treatment.
The advent of the NeuroTrack™ assay provides a solution to these issues. The combination of NeuroTrack software and the IncuCyte™ live-content imaging platform measures the neurite dynamics of the live cells without the need for a fluorescent label.
Under physiological conditions, the IncuCyte captures non-perturbing phase-contrast images of neuronal cultures for 5 days to 3 weeks (Figure 1, top image). These images are then analyzed by the NeuroTrack software to generate data on neurite dynamics (Figure 1, lower images).
The key advantage is the ability to gain insights into the kinetic effects of the treatment of interest as the data obtained from a culture is for a complete time course.
NeuroTrack can be used in conjunction with various neuronal model systems as it can quantify the robust processes of differentiated Neuro-2a and PC-12 cells and very fine neurites of iPSCs and primary cells. This article describes the validation of the NeuroTrack assay for medium-throughput testing in a 96-well format.
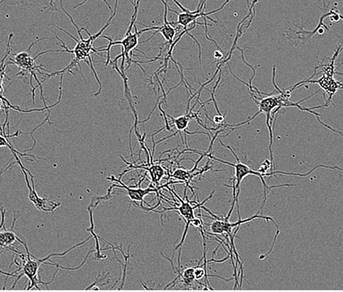
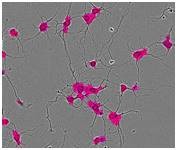

Figure 1. Segmentation masking of rat E18 cortical neurons. Phase image of neurons (top) after 5 days in vitro (DIV). Cell body cluster mask applied to image (bottom left). Neurite mask and cell body cluster mask applied to image (bottom right).
Approach and methods
Rat E18 cortical and hippocampal cultures
The day before the dissociation of tissues, WFI-grade water was used to rinse a 96-well plate from Techno Plastic Products for two times and the plate was then coated overnight with 150μL/well of 0.1mg/mL poly-d-lysine in WFI water.
The following day, the remaining poly-d-lysine was removed by rinsing the plate twice, followed by drying the plate in a biosafety cabinet. 100μL/well of 3.3μg/mL laminin was then applied over the poly-d-lysine coated plates, which were subsequently incubated for at least 1 hour at 37°C.
The dissociation of rat E18 cortical or hippocampal tissue held at 4°C was carried out within four days of dissection. Storage medium (Hibernate-E) was separated from the tissue and stored.
The tissue was treated with 2μg/mL papain in calcium-free Hibernate medium in a water bath at 37°C for 30 minutes. After the removal of the enzyme solution, the cells were coated with the saved storage media and triturated around 10 times with a firepolished, silanized Pasteur pipette.
The dispersed cells were then shifted to a 15ml conical tube and centrifuged at a speed of 1000rpm for 1 minute. After the removal the supernatant, the pellet was resuspended in Neurobasal medium at 37°C and supplemented with 2% B27, 0.5mM Glutamax, and with 25μM glutamate for hippocampal cultures.
A 40μm cell strainer was then used to filter the cell suspension, and the cells were then plated in coated plates at 8-10,000 cells/well of a 96-well plate (285 cells/mm2), in 150μL of supplemented media/well and incubated at 37°C, 5% CO2.
After plating for 18 to 24 hours, the media was replaced with 150μL/well of supplemented Neurobasal medium comprising of test compounds. 25μM glutamate was kept in the media for five days for hippocampal cultures.
The addition of glutamate was avoided if it is required to change the media. For cytochalasin D and Ro-31-8220 experiments, the vehicle used was DMSO at concentrations of 0.01%, a concentration earlier demonstrated to be nonperturbing.
Neuro-2a cultures
Cells were kept in F-12K medium supplemented with 5% FBS and incubated at 37°C, 5% CO2. Cells were differentiated by trypsinizing, pelletizing, and resuspending them in serum-free F12K. The cells were then plated in a 96-well TPP plate at 2,000 cells/well (57 cells/mm2) in serum-free F12K.
Newly seeded plates were placed in a 37°C incubator for 1 hour. The final FBS concentration was brought to 2% and the final all-trans retinoic acid concentration to the required concentration by adding differentiation medium to the wells. The vehicle used was DMSO at a concentration of below 0.1%.
Neuro-2a NucLight Cultures
A lentivirus encoding a puromycin resistant gene was used to stably transduce neuro-2a cells for clonal selection, and a nuclear restricted fluorescent protein was used to produce the Neuro-2a NucLight Red cell line or the Neuro-2a NucLight Green cell line.
The addition of 0.5μg/mL puromycin to the growth medium (F-12K, 5% FBS) ensured homogeneity of the culture. Puromycin was not included to the differentiation media. Here, Neuro-2a NucLight lines were treated as the parent line.
PC-12 cultures
Cells kept in DMEM supplemented with 10% horse serum and 5% FBS were incubated at 37°C, 5% CO2. Cells were differentiated by trypsinizing, pelletizing and resuspending them in serum-free DMEM.
The resultant cells were then plated in a 96- well TPP plate at 2,000 cells/well (57 cells/mm2) in serum-free DMEM. Newly seeded plates were then placed in a 37°C incubator for 2 to 4 hours.
The final horse serum concentration was brought to 1% and the final NGF concentration to the prescribed concentration (10-100ng/mL) by adding differentiation medium.
iCell® Neuron cultures
On the day of plating, WFI-grade water was used to rinse a 96-well TPP plate for two times and then the plate was layered with 150μL of 0.1 mg/mL poly-d-lysine in WFI water and incubated for two hours at room temperature.
After rinsing the plate for two times to eliminate the remaining poly-d-lysine, it was dried in a biosafety cabinet. The next step was coating the plate with 100μL/well of 3.3 μg/mL laminin and incubating at 37°C for at least one hour.
The laminin solution was isolated without any delay before seeding cells. The preparation of cells was carried out as outlined in the iCell ® Neuron User Guide. The resultant cells were then seeded at densities between 8k and 25k cells/well.
200μL/well of iCell ® Neuron Complete Maintenance Media then replaced the existing media after 18 to 24 hours of plating. After cells of interest had been seeded, vessels were positioned in an IncuCyte to capture phase-contrast images.
The images were investigated every 4 to 6 hours over the course of the assay. A 20x objective was used to image E18 cortical, hippocampal and iCell ® neurons. Neuro-2a cells and PC-12 cells were well of a 96-well plate at 20x, and 4 images per well at 10x.
Data quantification and analysis
An HD phase image of neurons is analyzed by the NeuroTrack software in two steps:
- Segmentation of cell bodies from background is carried out on the basis of texture and/or brightness, followed by masking the cell bodies as cell body clusters (Figure 1, lower left). The next step is normalizing the number of clusters and the total area of clusters to image area.
- Linear features are determined based on brightness and width, and are masked as neurites (Figure 1, lower right). The next step is normalizing the total neurite length and the total number of branch points to the image area.
It is possible to refine and filter the segmentation mask to customize the mask to the specific cell type. The automated software analysis generates the following list of metrics for every timepoint of a NeuroTrack assay:
|
NeuroTrack™ Metrics
|
|
Neurite length
Total neurite length/mm2
Neurite length/cell body cluster count
Neurite length/cell body cluster area
Cell body clusters
Count/mm2
Area/mm2
|
Branch points
Total branch points/mm2
Branch points/cell body cluster count
Nucleus count (for cells with a nuclear
fluorescent label)
Nucleus count/mm2
Neurite length/nucleus count
Branch points/nucleus count
|
Using these metrics, biologically relevant processes like neurite initiation, neurite branching, extension, and loss of neurite length caused by retraction or disintegration are quantified. Quantifying the count and area of the cell body clusters provides measurement methods for cell body size or proliferation and two metrics for normalization.
It is possible to normalize NeuroTrack metrics to a fluorescent nuclear count when a red or green nucleus is present in the model system. One such model system is Neuro-2a NucLight™ cells from Sartorius.
It has a fluorescent nucleus that can be observed within the NeuroTrack application, automatically producing statistical data such as standard deviation and S.E.M. for user-defined replicates.
Results and discussion
Assay Flexibility
Five different cell types were used to validate the NeuroTrack processing algorithm:
- Primary E18 rat cortical neurons
- Primary E18 rat hippocampal neurons
- iPSC-derived neurons (iCells® from Cellular Dynamics International)
- Neuro-2a cells
- PC-12 cells
For each cell type, an optimized processing definition was utilized in experimental assays. Monitoring the NeuroTrack™ masks at early, middle and late timepoints corroborated the effective masking of neurite dynamics.
Figure 2 presents the HD phase images and the corresponding NeuroTrack masks, showing cells at mid-to-late timepoints in the assay at the same scale.
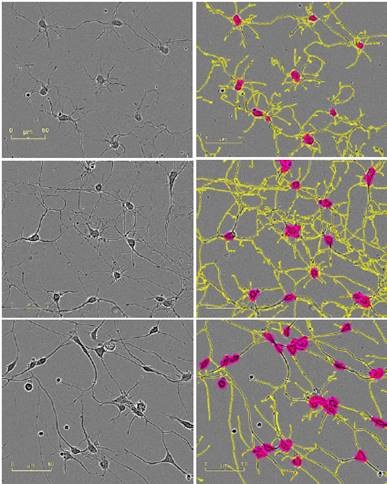
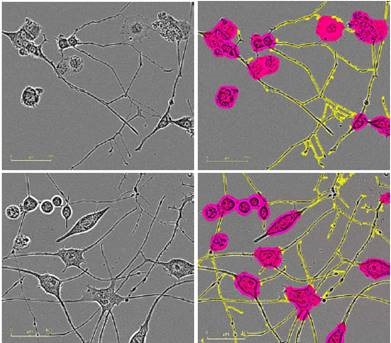
Figure 2. Five neuronal model systems imaged in an IncuCyte in phase (left) are successfully masked by NeuroTrack (right). From top in descending order: E18 rat cortical neurons, 4.5 days post plating; E18 rat hippocampal neurons, 4.5 days post plating; iPSC-derived neurons, 3.5 days post plating; Neuro-2a cells, 3.5 days post treatment with 20μM all-trans retinoic acid in 2% FBS; PC-12 cells, 5 days post treatment with 50ng/mL NGF in 1% horse serum. Raspberry: cell body clusters; yellow: neurites.
Assay variability and validation
The NeuroTrack assay shows the low variability essential for medium-throughput screening, thanks to precise quantitation and optimized cell culture methods. A 96-well plate of E18 rat cortical neurons plated at 8,000 cells/well on polyd-lysine was used to examine the intra-assay variability of NeuroTrack.
From a five-day time course charting neurite length/cell body cluster count, the coefficient of variation was determined to be maintained below 10% throughout the whole assay (Figure 3A). Moreover, the undesirable edge effects (Figure 3B) were determined by analyzing the 96-well plate format.
Neurite length/cell body cluster did not vary significantly between the cells plated in edge wells and cells plated in the center during the five-day time course. Power analysis of rat cortical neurons reveals that it is possible to reach more than 80% power using 6 wells per condition and 9 images/well captured at 20x to quantify a 10% change in neurite outgrowth/cell body cluster count.
This result confirms the amenability of the NeuroTrack assay to screening protocols and its ability to reduce reagent consumption and provide rapid results for cell function analyses.

Figure 3. Intra-assay variability. A) Intra-assay variability of a 96-well plate of rat cortical neurons plated on poly-d-lysine at 8,000 cells/well. Blue shows neurite length, error bars represent standard deviation (left y-axis). Green shows coefficient of variation (right y-axis). B) Edge effect analysis of the same plate. Error bars are standard deviation. N=96.
The NeuroTrack assay was validated by comparing the data from live-cell NeuroTrack analysis of neurite outgrowth to the data obtained from fixed, end-point analysis. Three 96- well plates from the same E18 rat cortical prep were specifically seeded at 8,000 cells/well and each plate was imaged in ZOOM at various timepoints post-plating (18, 90, and 162 hours).
Neurite length/mm2 was then measured by quantifying the images with NeuroTrack. Plates were displaced from the incubator without any delay after imaging in ZOOM, and fixed and immunostained for β-tubulin for marking the neurite structures.
An Image Xpress Micro high-content screening system was then used to image the cells and neurite length/mm2 was quantified using MetaXpress software. Figure 4 shows the comparison of the quantification from NeuroTrack and MetaXpress software.
NeuroTrack’s automated software measured neurite length in unfixed cells during a 6.5 day assay with a sensitivity statistically equivalent to the fixing/immunostaining method measured by the MetaXpress software, at the three different time points.
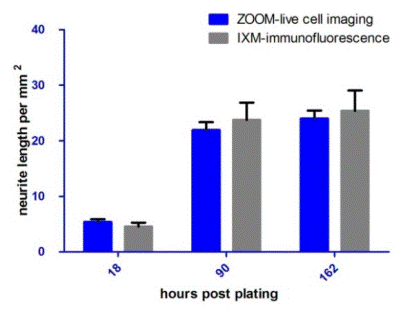
Figure 4. NeuroTrack ZOOM vs. High Content Imaging and Analysis. Neurite length (mm/mm2) of cells imaged and analyzed by NeuroTrack ZOOM and then immediately fixed, immunostained for β-tubulin, and imaged and analyzed by Image Xpress Micro. The two data sets are statistically indistinguishable at each time point. Error bars represent standard deviation. N=6.
NeuroTrack™ applications
Time-dependent effects of cytochalasin D on neurite length
The alkaloid cytochalasin D is recognized as a potent inhibitor of F-actin polymerization.1 When cytochalasin D is present in higher concentrations, multiple axon-like structures are shown to be rapidly developed in rat primary hippocampal cells within 24 hours after treatment.2,3,4
However, in the cited studies, endpoint assays were utilized and therefore, neurite length was not measured beyond the 24-hour time point. A more complete time course of the effects of cytochalasin D was investigated by treating rat cortical neurons at 1 day in vitro (DIV) with three different concentrations of cytochalasin D (Vehicle: 0.01% DMSO), with monitoring of neurite length per cell body cluster count every six hours.
At 24 hours post-treatment, the results obtained clearly showed the increase in neurite length as reported in the literature. ~33% greater neurite length/cell body cluster was observed for cells treated with 1μM cytochalasin D (Figure 5).
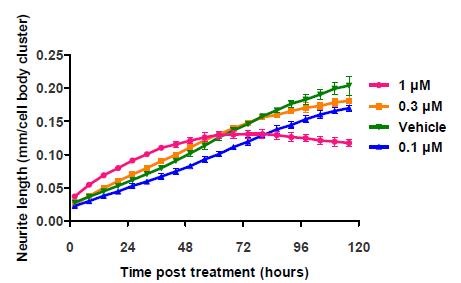
Figure 5. Application of cytochalasin D to E18 rat cortical neurons shows a fluctuation of neurite length at the highest concentration. The literature cited only reports data taken at and before the 24 hour time point. Data points represent mean ±standard deviation. N=6.
However, the initial rate of increase decreased after 48 hours, and the length started to reduce by 96 hours. These results were corroborated by the images of the cortical neurons, which revealed that the loss of neurite length was due to the cell death caused by high concentration of cytochalasin D over time (Figure 6).
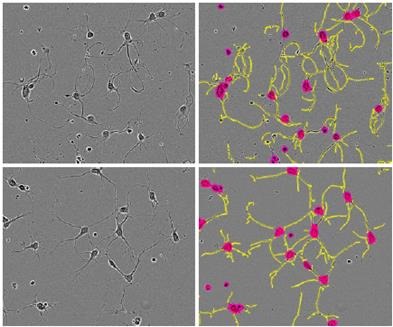
Figure 6. Phase images of E18 rat cortical cells treated with 1 μM cytochalasin D (top) or vehicle (bottom) and their corresponding NeuroTrack™ masks. Taken from t=24 hours post application, when there is a 33% greater neurite length/cell body cluster in the 1μM treated cells. Note the smoother, curlier neurites in cytochalasin Dtreated cells.
The images also revealed that cytochalasin D in lower concentrations did not lead to a significant initial burst of axon formation nor cause a strong toxic effect. However, similar morphological responses were observed for cells treated with all concentrations of cytochalasin D.
Smooth and curling neurites were observed when compared to control cells (Figure 6, top row). This analysis was performed carried out for two times with the same kinetic response (data was not presented here).
Neurotoxicity predicted by neurite length measurement
Neurite length is useful data to quantitatively measure the neurotoxicity. This was demonstrated by utilizing a well-characterized PKC inhibitor, Ro-31-8220, and plating E18 rat cortical neurons at 10,000 cells/well and treating them with the given concentrations of Ro-31-8220 at 1 DIV.
In comparison with the vehicle control (0.01% DMSO), the neurite length was reduced in a concentration-dependent manner when Ro-31-8220 was added to cortical neurons (Figure 7). The data was validated by the images of the cells by revealing disintegrated neurites and shrunken cells in treated wells (Figure 8).
The low variability shown in the analysis suggests that the 96-well assay is suitable for medium-throughput screening applications.
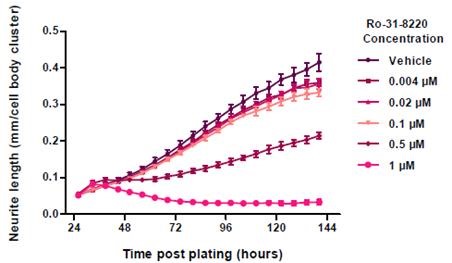
Figure 7. Neurite Length in response to Ro-31-8220 treatment. Data points represent mean ±standard deviation. N=6.
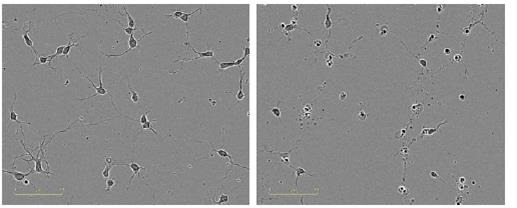
Figure 8. Qualitative observation of neurotoxic effect of Ro-31-8220. Images taken in ZOOM at 20x, 24 hours post treatment (48 hours post plating). Left, vehicle control; right, 1μM Ro-31-8220.
Conclusion
The live-cell imaging platform of the IncuCyte in combination with NeuroTrack software offers a label-free, live-content imaging assay capable of kinetically quantifying neurite dynamics in living cultures.
This highly sensitive method can determine pharmacological and genetic manipulations that affect formation, elongation, and loss of length of neurites. It can be used for a variety of neuronal culturing methods and is amenable to medium-throughput screening.
The imaging strategy enables validation of all quantitative data by observing morphological changes, thereby facilitating uniquely thorough analyses to be performed in a single assay.
The image analysis software can be used to measure different types of cells, ranging from large immortalized cell lines and primary neurons to iPSCs. Neurite dynamics can be rapidly optimized and quantified, thanks to the intuitive user interface.
Kinetic measurements of neurite length, cell body clusters, and branch points provide a complete picture of a highly dynamic process, which cannot be thoroughly understood by single time point data alone. This ability eliminates the need for arbitrarily defined endpoints.
Minimal user labor and low well-to-well variability make the assay robust enough for medium throughput screening in a 96-well format
The NeuroTrack assay can be multiplexed by the users. It is possible to use fluorescent markers, such as mix-and-read endpoint dyes or nonperturbing fluorescent protein reporters, to allow for IncuCyte fluorescent measurement of real-time proliferation, neurotoxicity, or cell viability.
It is possible to verify all data and time points by investigating individual images and/or time-lapse movies. Monitoring cell morphology offers additional validation and knowledge about the biological effect of treatment groups.
Acknowledgements
Produced from materials originally authored by Patricia M. Garay, Nevine Holtz, Vince Groppi and Lauren McGillicuddy from Sartorius, Ann Arbor, Michigan.
References
- Flanagan, M.D. and Lin, S. Cytochalasins block actin filament elongation by binding to high affinity sites associated with F-actin. J. Biol. Chem. 1980, 255(3):835-838.
- Bradke F. & Dotti C.G. The role of local actin instability in axon formation. Science 1999. 283(5409)1931–1934.
- Kunda P., Paglini G., Quiroga S., Kosik K., & Caceres A. Evidence for the involvement of Tiam1 in axon formation. J. Neurosci. 2001, 21(7)2361–2372.
- Ruthel G. & Hollenbeck P.J. Growth cones are not required for initial establishment of polarity or differential axon branch growth in cultured hippocampal neurons. J. Neurosci 2000, 20(6):2266–2274.
Sartorius

Sartorius is a leading international pharmaceutical and laboratory equipment supplier. With our innovative products and services, we are helping our customers across the entire globe to implement their complex and quality-critical biomanufacturing and laboratory processes reliably and economically.
The Group companies are united under the roof of Sartorius AG, which is listed on the Frankfurt Stock Exchange and holds the majority stake in Sartorius Stedim Biotech S.A. Quoted on the Paris Stock Exchange, this subgroup is comprised mainly of the Bioprocess Solutions Division.
Innovative Technologies Enable Medical Progress
A growing number of medications are biopharmaceuticals. These are produced using living cells in complex, lengthy and expensive procedures. The Bioprocess Solutions Division provides the essential products and technologies to accomplish this.
In fact, Sartorius has been pioneering and setting the standards for single-use products that are currently used throughout all biopharmaceutical manufacturing processes.
Making Lab Life Easier
Lab work is complex and demanding: Despite repetitive analytical routines, lab staff must perform each step in a highly concentrated and careful way for accurate results.
The Lab Products and Services Division helps lab personnel excel because its products, such as laboratory balances, pipettes and lab consumables, minimize human error, simplify workflows and reduce physical workloads
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.